



Issue:May 2014
FORMULATION DESIGN - Formulation of Poorly Soluble Drugs: A Modern Simulation-Based Approach
ABSTRACT
Leading companies in other industries have shown that designing for quality and manufacturability from the earliest stages can positively impact the outcomes for a product. In addition, predictive modeling and directed product analyses can significantly reduce time-to-market and overall product costs.
To enable such approaches in pharmaceutical formulation development, there is a strong need for deeper mechanistic understanding to support efficient and robust formulation activities for poorly soluble molecules. To meet this need and the demands of clients seeking solubilization solutions, Agere is taking a modern approach to solubilization formulation development that incorporates Quality-by-Design (QbD) principles at the preclinical stages of the drug development process. We have developed a platform, Quadrant 2™, that is based on a comprehensive understanding of multiple proven solubilization technologies coupled with materials science and a targeted application of molecular modeling. The primary advantage of this approach is that it enables a faster, more informed formulation development process that promises to minimize costly iterations and reformulations as drug products proceed into the clinic.
The following presents a case study in which a model BCS Class II compound, dipyridamole, was evaluated as an amorphous dispersion using molecular modeling combined with experimental data. All work described was conducted using Agere’s Quadrant 2 platform.
INTRODUCTION
Solubilization formulation design has traditionally been a stage of product development that takes place long before QbD guidelines are applied. One reason for this is that pharmaceutical unit operations naturally lend themselves to the use of well-established tools, such as design of experiments. A second reason is that prediction of the behavior of materials is complex, and no comprehensive materials science modeling tools had been developed directly related to drug delivery. To address this need, Agere has undertaken the development of a set of tools that combine physical measurements and simulations to fully understand the fundamental materials science of formulation design.
IN SILICO MODELING
The in silico modeling phase of Quadrant 2 is a crucial first step in dispersion development and manufacture. It provides the context and constraints needed to screen candidate excipients and solvent systems for a given chemical entity. Modeling starts at the molecular-level and progresses through thermodynamic and kinetic analyses. Each stage of analysis is designed to explore and refine interactions and relationships among APIs, excipients, and solvents resulting in a pre-optimized formulation. This powerful tool leads to informed selection of finalist systems prior to full laboratory evaluation.
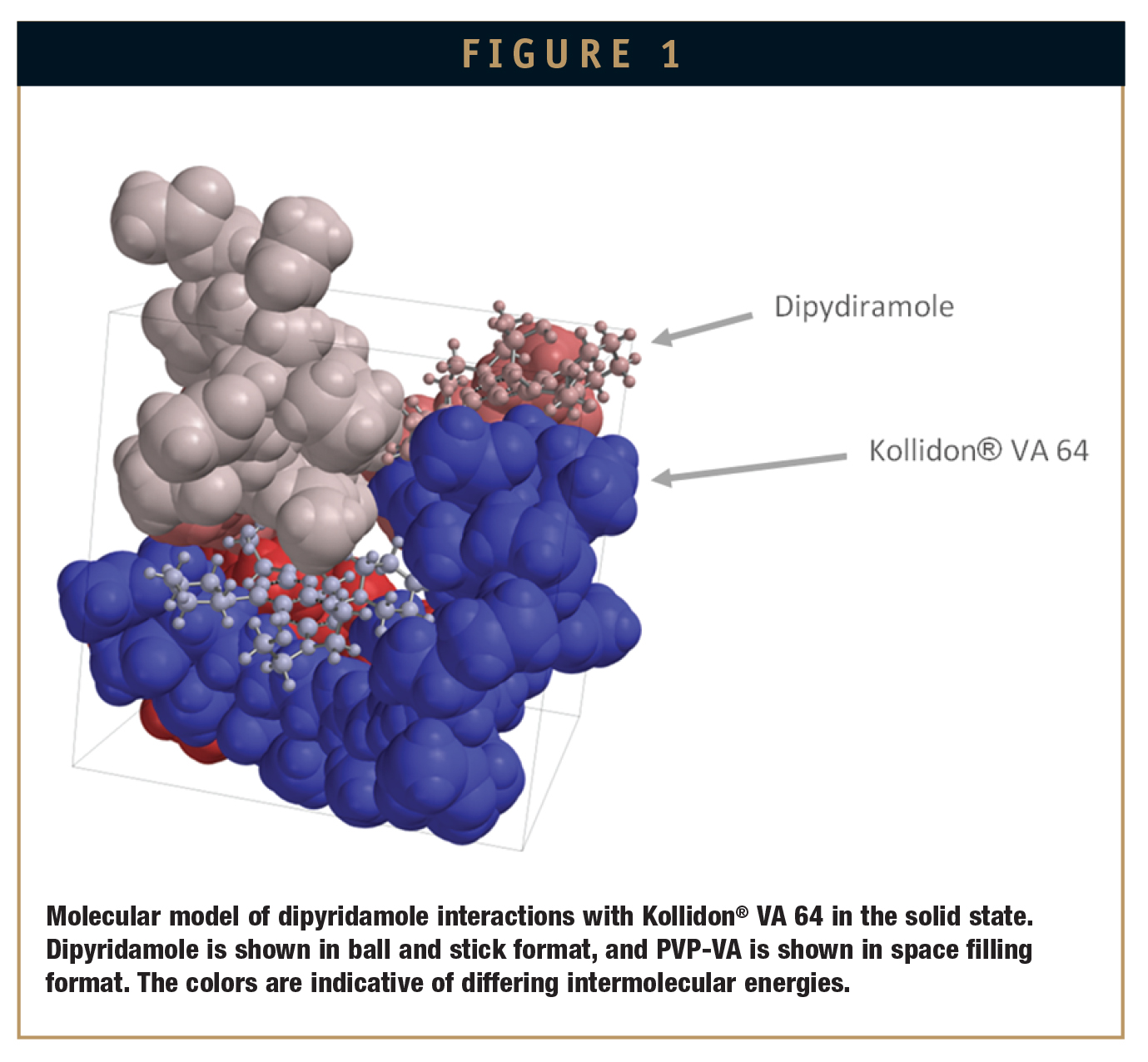
A representative molecular model of dipyridamole with Kollidon® VA 64 (PVPVA) as the dispersion polymer is shown in Figure 1. The modeling of dipyridamole:PVPVA dispersions show strong non-covalent interactions between the hydroxyl groups of dipyridamole (H-bond donating) and carbonyl groups of PVP-VA (H-bond accepting). In addition, it is informative to visualize the molecular environment of the drug in the polymer matrix. Specifically, comparison of the molecular configuration of the drug in its crystalline form (not shown) with that in the polymer clearly demonstrates there are significant steric conformation changes between the two states. In fact, it is these specific changes that are, in large part, responsible for the free energies of mixing that ultimately lead to the bulk properties of the solid dispersion.
INFRARED SPECTROSCOPY
Fourier transform infrared spectroscopy (FTIR) is a powerful technique for the study of molecular interactions. In addition, the vibrational energies measured experimentally are directly comparable to those calculated from high-level quantum mechanical simulations. To aid our understanding of the drug-polymer interactions, FTIR spectra were collected for dipyridamole, PVP-VA, and dispersions thereof.
Spectra were recorded for pure amorphous dipyridamole, PVP-VA, and spray-dried dispersions of dipyridamole:PVP-VA (10:90, 50:50, and 70:30 wt%) on a Nicolet iS10 FTIR spectrometer in the range from 4000 to 500 cm-1 at 25°C. Select regions of the spectra are shown in Figure 2.
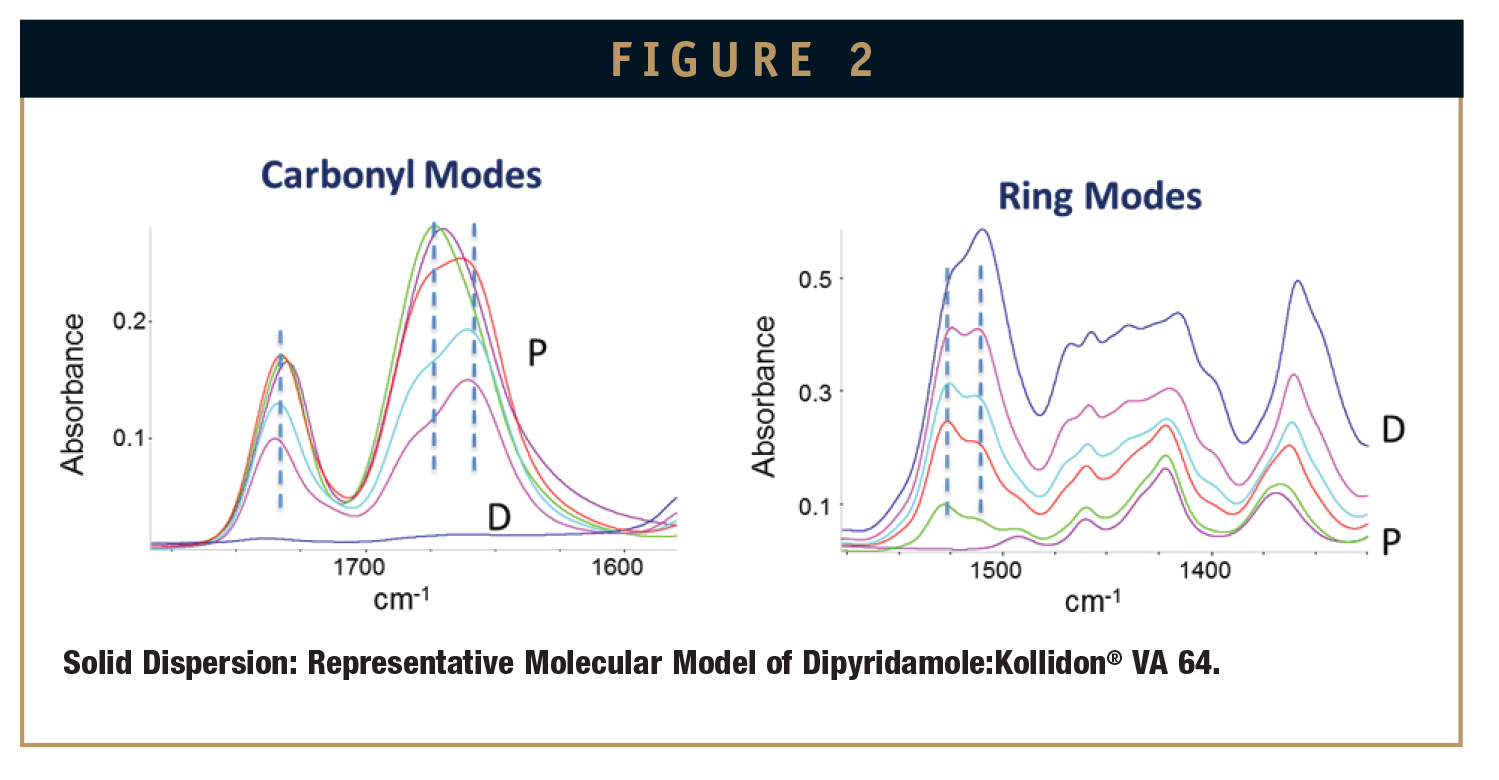
PVP-VA has no hydrogen bond-donating capability; however, there are two types of hydrogen bond-accepting carbonyl groups on the pyrrole ring, and one on the acetate group. The characteristic vibrational peaks are centered at 1729 cm-1 and 1670 cm-1. Dipyridamole contains four hydroxyl (-OH) groups that can act as hydrogen-bond donors, and these –OH-stretching modes appear as a broad band spanning from ca. 3000 to 3600 cm-1 (data not shown). The ring modes of dipyridamole span the region from ca. 1500 to 1525 cm-1 with peaks centered at 1510 cm-1 and 1524 cm-1.
As shown in Figure 2, IR spectra of spray-dried dispersions of dipyridamole:PVPVA show distinct spectral changes with respect to the pure components (D indicates pure amorphous dipyridamole, and P indicates pure PVP-VA). They were identified in two different regions associated with the carbonyl modes of PVP-VA and the ring modes on dipyridamole.
Upon interaction with dipyridamole, the carbonyl band of PVP-VA centered at 1670 cm-1 is split into two discernible bands with peaks at ca. 1660 cm-1 and 1680 cm-1, and the band at 1729 cm-1 shifts slightly to 1738 cm-1. Concomitantly, upon interaction with PVP-VA, the hydroxyl bands of dipyridamole undergo a shift from a maximum of 3260 cm-1 (pure dipyridamole) to 3440 cm-1 (70% dipyridamole) [data not shown]. The ring modes of dipyridamole also undergo a slight shift and change in intensity upon interaction with PVP-VA. Molecular modeling suggests that as the drug becomes more dilute in the polymer, the intra-molecular hydrogen bond on the dipyridamole ring system rotates outward and hydrogen bonds preferentially to a carbonyl group on PVP-VA. The combined spectral changes observed in the spray-dried dispersions are indicative of hydrogen bonding interactions that occur between the hydrogen bond acceptor groups of PVP-VA and the hydrogen bond donor groups on dipyridamole.
IR spectra were also calculated in silico using density functional theory (DFT) and used to assign the fundamental modes of vibration for the dipyridamole, PVP-VA, and specific drug-polymer interactions. With these methods, we are able to reasonably predict the major features of the experimental IR spectra.
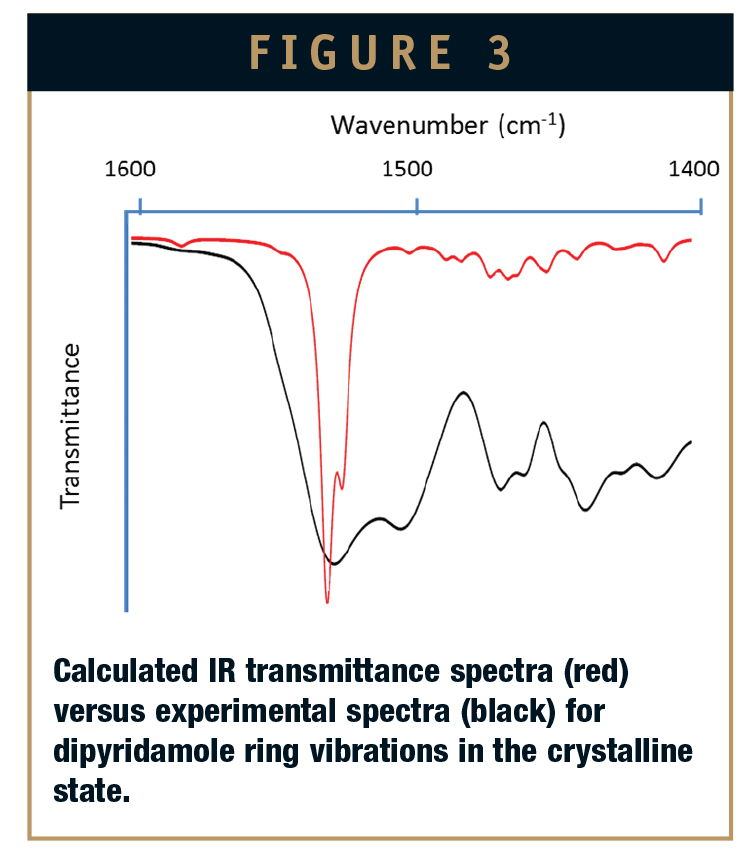
A representative IR transmittance spectra for crystalline dipyridamole is shown in Figure 3, illustrating the region of the oscillating ring modes. The calculated spectra are shown in red, and the experimental spectra are shown in black. Peak positions of the calculated vibrational modes allow assignment of the fundamental vibrational modes in the experimental data. Note that exact fits of experimental to calculated spectra are not expected due to a number of factors in the quantum mechanical calculations. Specifically, the calculated spectra contain only the 3N-6 fundamental vibrational modes of the molecule in vacuum. In comparison, the experimental spectra unavoidably have numerous intermolecular interactions that result in additional vibrational energies and intensities. In addition, the calculated vibrational spectra do not include hot bands or the various allowed combination and difference transitions that are present in the experimental results. To address these differences, we are developing methods to enhance the modeling and predictive aspects of the calculations.
FUTURE DIRECTIONS
Agere’s approach in developing a methodology for drug formulation design involves expanding Quadrant 2 to encompass a comprehensive set of in-depth tools that combine physical measurements with a variety of calculation modules to fundamentally understand and predict drug and polymer interactions.
As can be seen, the use of quantum mechanical methods to elucidate the fundamental properties of drug and polymer interactions has particular utility. Statistical mechanics methods are then used to transform the molecular-level interactions into macroscopic-level interactions via the partition function. In conjunction with the modeling, highly directed experimental measurements are used to confirm and refine the predictions from the simulations. Using this approach, we are able to not only efficiently select and develop an optimum set of materials for use in a solid dispersion, but also able to inform why these formulations are most optimal.
To view this issue and all back issues online, please visit www.drug-dev.com.

Dr. Sanjay Konagurthu is Senior Director of Formulation Sciences at Agere Pharmaceuticals, Inc. and has over 15 years of experience managing the development of compounds from the discovery interface through clinical manufacturing. His expertise spans a broad spectrum of therapeutic areas, all involving formulation and process development of novel drug delivery technologies. With extensive experience in oral solid dosage forms, including solubility enhancement and modified release, Dr. Konagurthu has performed and managed the formulation and process development for a broad range of NCEs and lifecycle management of marketed products. He has 7 patents/patent applications to his name, and has authored more than a dozen publications. Dr. Konagurthu earned his Bachelors of Technology from the Indian Institute of Technology (IIT), Madras, and his PhD from the University of Colorado, Boulder, both in Chemical Engineering.

Alexander McVey is a Research Scientist at Agere Pharmaceuticals, Inc. He joined the company in 2012 upon completing his MS in Chemistry from the University of Oregon, where he specialized in polymers and coatings. In his current position, he is involved in research and development focusing on analytical support and formulation development for spray-dried dispersions, other amorphous dispersion technologies, and immediate-release solid dosage forms. Past positions include QC chemist in Quality Control at Agere, and Chemist at Absorption Corp. (now J. Rettenmaier & SÖhne Group). Alexander completed his undergraduate work at Western Washington University, where he earned his BS in Chemistry with a minor in Materials Science.
Total Page Views: 2710












