

Issue:March 2017
BIOMARKERS - FDA’s Design Control Requirements for Biomarkers in Drug Development
INTRODUCTION
The availability of validated biomarker-drug companion products will enable the molecular diagnostics and pharmaceutical industries to develop and rely on new genomic biomarkers in order to elucidate disease pathways, stratify patient populations, and monitor safe and effective use of these products for personalized therapeutic use. The use of companion biomarker with a particular drug application is stipulated in the instructions for use in the labeling of the product.
BIOMARKER CLASSIFICATION & QUALITY SYSTEM REGULATIONS
Biomarkers are in vitro diagnostic devices (IVDs) regulated under the FDA’s Medical Device Law. A biomarker IVD provides measurable features or performance characteristics that are objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention. Biomarkers measure disease presence or progression that defines unique disease pathogenesis or responses to therapy. Biomarker tests are intended to perform a clinical assessment, such as blood analyte levels used to monitor and predict health status in individuals or across populations so that appropriate therapeutic intervention can be applied. Biomarkers may be used alone or in combination to assess or monitor the health or disease state of an individual. From a molecular perspective, biomarker IVDs are developed using genomics/proteomics technologies. It is imperative to distinguish between disease-related and drug-related biomarkers. Disease-related biomarkers give an indication of the probable diagnosis in cancer patients. Drug-related biomarkers indicate whether a drug will be effective in a specific patient or how the patient’s body will process it. Biomarkers are also considered as the key to personalized medicine treatments that are individually tailored to specific patients for highly efficient intervention in disease processes. Biomarkers play a key role in design, discovery, and drug development. The availability of validated biomarkers will enable the pharmaceutical industry to bring revolutionary new medicines to market more quickly, safely, and less expensively.
BIOMARKER DESIGN CONTROLS
Design controls are a mechanism for bringing the design of certain class I and all class II and class III investigational devices under the umbrella of the good manufacturing practices (GMPs) of a corporate quality system. In 1996, the GMP requirements were revised to include the area of design control and have become a part of the Quality System Regulations (QSR) with which all medical device manufacturers must comply. It is incumbent on the manufacturer to demonstrate compliance with the QSR and, as of 1996, with design control requirements as well. Once the product definition and regulatory strategy have been prepared, IVD medical device developers must work to comply with the design control provisions of the QSR as the device development process moves forward. The QSR is the medical device equivalent of the pharmaceutical current good manufacturing practices (cGMPs). The QSR, unlike cGMPs, also regulates the IVD device development process via its design control provisions (21 CFR 820.30), which describes the IVD device developer’s requirements under the design control provisions of the QSR. Design controls are an integrated set of management practices (policies, processes, and procedures), which are applied to control design activities while assessing quality and correcting errors through a reiterative device process control. Once management has determined that the device is feasible and the decision has been made to transition from research to clinical applications, the design control process begins. As such, the device application becomes part of a corporate quality system (Figure 1).
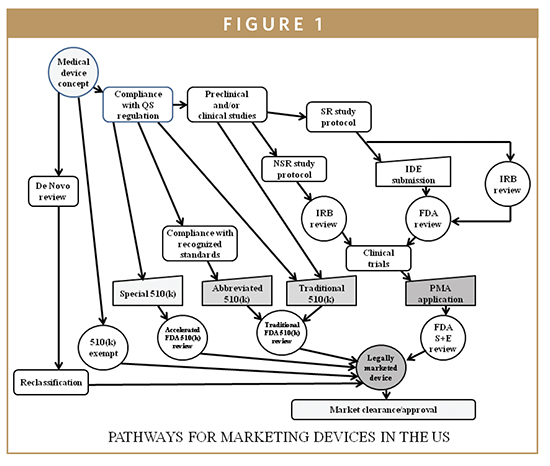
DESIGN CHARACTERISTICS OF BIOMARKERS
The FDA requires the consideration of human factors during the development of medical devices. In 1996, the FDA issued guidance, “Do It By Design-An Introduction to Human Factors in Medical Devices,” which established design requirements to avoid so-called user errors.1 Most recently, the agency introduced those concepts of human reliability to drug and biotechnology manufacturers. To elaborate this concept by frequency and by significance, one must differentiate between error (mistakes) and defects (also known as nonconformance). It is difficult for regulated firms to recall products because there were human errors during the manufacturing process. In some situations, the firm’s quality system is unable to detect the human error and the nonconformed device distribution becomes an adulterated item. Human error can be defined as a departure from acceptable or desirable practices on the part of an individual, which results in unacceptable or undesirable outcome. Human factors include designing machines, operations, and work environments to match human capabilities, limitations, and needs. In those situations in which an operator does not properly execute a manufacturing step, human error can be avoided by using risk management tools described in ISO 14971-Application of Risk Management to Medical Devices [Hazard and Operability Study (HAZOP) and Hazard Analysis and Critical Control Point (HACCP)]. HACCP for medical devices is designed to prevent and/or control device safety and performance (Figure 2). A trained HACCP team is helpful for monitoring the IVD device production processes from raw materials, components receiving, manufacturing controls, distribution, and use by the customer. Since HACCP focuses primarily on the production process, it is also assumed that the design is complete, and the manufacturing system is already in place; however, HACCP, and/or the corrective and preventive action plan, may indicate the need to revise the design and/or the manufacturing process, subject to compliance with change control provisions of the QSR.2,3
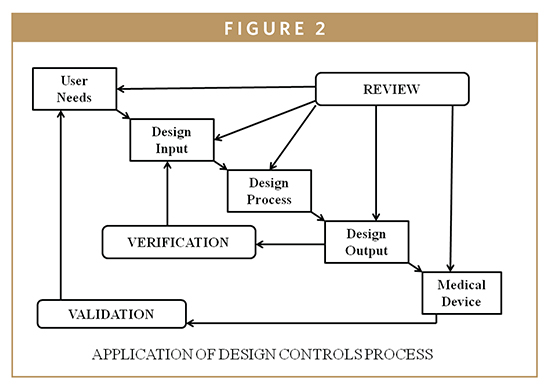
QUALITY SYSTEMS & GMPS FOR BIOMARKER APPLICATIONS
The QSR regulates both the IVD device development and the manufacturing process for all class II and class III devices from the beginning of the design engineering development phase until commercial use and post-marketing surveillance. The IVD Biomarker Total Product Life Cycle (TPLC) interconnects all developmental aspects, manufacturing, and commercial use of products along with quality monitoring via design control requirements (Figure 3). The QSR also covers the manufacturing process for many class I devices. The goal of the QSR is to create a self-correcting system that reliably produces robust IVD device designs and production methods, ensuring these devices perform in a manner consistent with their intended use. Much of the information that is included in a 510(k) or PMA is taken from the Design History File (DHF), prepared as a result of the design control requirements of the QSR. Once a device is marketed, the corrective and preventive action (CAPA) provisions of the QSR are closely related to compliance with the MDR regulation. An additional feature of the QSR is that it follows the basic principles of the international medical device standard, ISO 13485, which is advantageous to enable device firms to sell their devices internationally to maintain quality system commonalities for most design and production-related activities. In most cases, the QSR system requires more extensive documentation than ISO 13485. The system requires documentation of specific activities of the development process, and documentation of specific evaluations and procedures of the manufacturing and quality processes. Frequently, the FDA investigators will follow the quality system inspection technique (QSIT) when inspecting a medical device facility (Figure 4). This process breaks the QSR compliance into four main modules and four satellite modules, some of which may not be applicable to all device firms. Generally, the FDA investigator will choose a subset of those modules and determine the device-specific compliance with QSR. This means that not every system’s modules are reviewed during a QSIT inspection; however, this approach does yield a general assessment of the QSR compliance. Many IVD device firms rely on various customer-oriented feedback loops and accountability of the process. This approach can be useful by reducing time to market and by reducing the number of field corrections and recalls contributing to increasing customer satisfaction and IVD device’s safety and effectiveness. The FDA’s guide to Inspections of quality systems provides instructions for conducting IVD device QSIT/GMP inspections.4-7
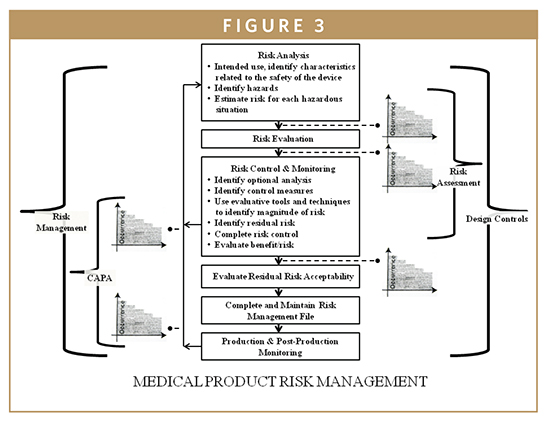
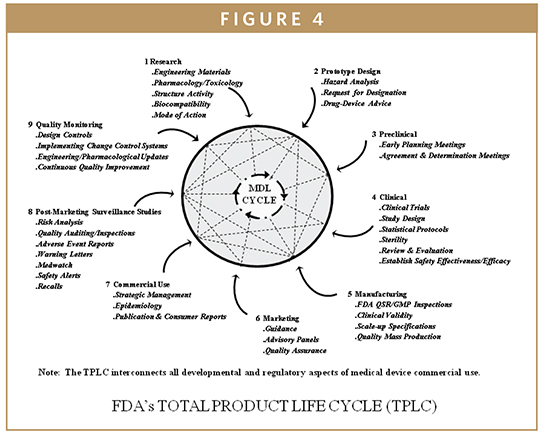
BIOMARKER DESIGN CONTROL COMPONENTS
The essential components of design controls stretch from planning through design transfer (from development, manufacturing to end user). Also, it is essential to maintain existing designs. These controls apply to all class II and class III medical devices and a small number of class I devices. The main purpose of the design control components are to establish and maintain procedures to control the design of the IVD device and maintain the intrinsic quality of the device in order to ensure that specified design requirements will meet user needs, the device’s intended uses, its specifications, safety and effectiveness, and reduction in recalls. These design components are developed in a reasonable manner in compliance with the firm’s existing design control standard operating procedures (SOPs). Design controls are closely linked to many other QSR components, and the entire quality system must work together to build and maintain the intrinsic quality of the IVD device. The device firms must prepare and follow SOPs that comply with the regulations and that fully describe how the firm will meet all relevant regulatory requirements. All the relevant activities must be fully documented in the firm’s design history file (DHF).
ELEMENTS OF DESIGN CONTROL REQUIREMENTS
The design control regulations require each manufacturer to establish and maintain procedures for the following:
-Design and development planning
-Design input
-Design output
-Design review
-Design verification and validation
-Design transfer
-Design changes
-Design history file
Medical devices, including IVD biomarkers that are intended for clinical use, are required to be cleared through the FDA’s premarket notification 510(k) or premarket approval (PMA) processes. Biomarkers, in comparison with other types of medical devices, are unique in the area of device regulation because they can be reviewed under specific labeling requirements (21 CFR 809.10). This regulation delineates the format and content of the information that the manufacturer of biomarkers must provide to the user (information required to support biomarker test system’s labeling in terms of product’s use and expected performance).
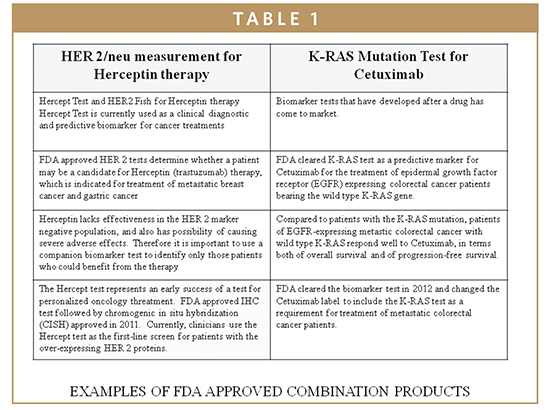
The QSR requires manufacturers of biomarkers to establish and maintain procedures that control the design of the biomarker system in order to ensure specified design requirements are met. The intrinsic quality of devices, including their safety and effectiveness, is established during the design phase. Thus, appropriate design controls are observed and maintained during preproduction stages of development so that finished devices are safe and effective for their intended clinical uses (Table 1).
Risk analysis must be conducted for the majority of medical devices and IVDs subject to design control requirements. Design deficiency can be a major cause of quality problems. The main goal of establishing a design control plan is to address the impact of design concepts as they relate to clinical utility of biomarkers in order to enhance benefits while reducing risks. The key element of the design output is the preparation and transfer of all documentation that is required to manufacture the biomarker product with consistent built-in quality. The documents usually include evidence of manufacturing equipment qualification, process flow diagrams, protocol describing a set of manufacturing procedures, standard operating procedures for release specifications, and instructions. These documents are part of the Device Master Record (DMR), which provides the basis for GMP audits by the FDA.
FDA QUALITY SYSTEM INSPECTION TECHNIQUE (QSIT)
The FDA’s QSIT approach to inspection is derived from seven sub-systems described in the QSR (21 CFR, Part 820). Four primary areas are the main focus of inspection: management controls, design controls, corrective and preventive actions (CAPA), and production and process controls. The remaining three subsystems are covered via “linkages” within the QSIT inspection subsystems. The QSIT review includes both a broad review of whether the firm has procedures in place that meet the general QSR requirements and a closer detailed review of specific device technology applications and records to verify that the requirements have been implemented in actual production, design, and daily quality assurance situations described in the premarket applications (510(k) or PMA).
MOLECULAR BIOMARKERS
Molecular biomarkers have been cleared by the FDA using acceptable platforms based on applied genomics and proteomics principles. Molecular biomarkers incorporate useful techniques that are complementary to routine testing within clinical lab disciplines (clinical chemistry, hematology, microbiology, virology, and immunology). The development of molecular diagnostic tests are based on the type of application for which the test is designed and intended for particular use. The development can be based on analysis of the genetic material or its’ products (ie, an examination or detection of DNA, of DNA adducts, or of RNA or the proteins produced). These IVD tests are generally based on an analyte, such as DNA, chromosome, protein, or other gene products, to detect mutations, karyotypes, disease-related genotypes, and phenotypes intended for clinical uses. Such uses include diagnosis, monitoring, prognosis, identification of carriers, or prediction of disease risks.3,5,6 Recently, there has been heightened interest and development in the application of biomarkers in oncology, including the role of KRAS in CRC and other EGFR-associated cancers (Table 1). In patients whose tumors express the mutated KRAS gene, the KRAS protein, which forms part of the EGFR signaling pathway, is always “turned-on.” This overactive EGFR signaling means that signaling continues downstream even when the upstream signaling is blocked by an EGFR inhibitor, such as cetuximab (Erbitux), which results in continued cancer cell growth and proliferation. Testing a tumor for its KRAS status (wild-type vs mutant) helps to identify those patients who will benefit most from treatment with cetuximab.
The sensitivity and specificity of recently developed biomarkers have the ability to greatly enhance cancer detection and the drug development process. Additionally, biomarkers will enable clinicians to develop individualized treatment plans for their cancer patients; thus making it possible to tailor drugs specific to their patient’s specific tumor type. This will improve individual drug response rate with limited drug toxicity and costs associated with testing and related therapies.
SUMMARY
-Risk and hazard analysis activities are required as part of total product life cycle (TPLC).
-The standard for the application of risk management (ISO 14971) for medical products is part of TPLC.
-The risk management process covers risk analysis, risk evaluation, and risk controls through CAPA and design control requirements.
-Design control requirements described in this publication play a key role from biomarker design prototype, manufacturing process controls, and the finished product for user needs.
-The extent of testing and evaluation is proportional to the level of risks associated with the biomarker technology and intended clinical use.
-FDA reviews the “safety and effectiveness” of the biomarkers, and it is essential that any risks and hazards are mitigated to “acceptable” levels.
CONCLUSION
The FDA reviewers and field investigators evaluate the design control requirements and processes and make recommendations based on whether the manufacturer has the required checks and balances in place, and whether the manufacturer verifies and validates the implementation of the design control requirements in support of the sponsor’s 510(k) or PMA.
To view this issue and all back issues online, please visit www.drug-dev.com.
REFERENCES
1. Do It By Design: An Introduction To Human Factors in Medical Devices 1996. (http:www.fda.gov/cdrh/humfac/doit.html).
2. Aziz KJ. The FDA’s perspective on clinical laboratory devices: pre-market review and evaluation process. J Biomed Lab Sci. 1993;5:151-158.
3. Aziz KJ. Global harmonization and quality System applications for clinical diagnostics. Clin Chem Lab Med. 2001:39:76-78.
4. Aziz KJ. Clinical trial design: integrating biomarkers into drug development. J Clin Ligand Assay. 2007;30:10-19.
5. Aziz KJ. Clinical molecular biology: concepts and applications. Adv Clin Chem. 1996;32:39-72.
6. Aziz KJ. Tumor markers: reclassification and new approaches to evaluation. Adv Clin Chem 1999;33:169-199.
7. Aziz KJ. Drug monitoring: modern approaches to quality assurance; CRC Handbook of Analytical Therapeutic Drug Monitoring and Toxicology. 1997;1,335-349.

Dr. Kaiser J. Aziz completed a 30-year career with the FDA, where he was Director of Mechanics and Materials Science and Associate Director of Clinical Lab Devices. He worked with individual and industry organizations in both design and total product life cycle (TPLC) approaches to pre-market applications for medical devices, pharmaceuticals, and combination products. He has designed, developed, and presented numerous training programs for medical devices and drug development industries (FDA’s QSIT and HACCP Programs). His expertise includes the FDA’s Quality System Compliance, Clinical Research Assessment, Product Development, Guidance for Protocols for Pre-Clinical and Clinical Studies for US Premarket Approval Process. Dr. Aziz earned his MS from Michigan State University, his PhD from American University, and a Post-Doctorate in Health Services from the University of Southern California.
Total Page Views: 9825










