


Issue:January/February 2018
PARTICLE DESIGN TECHNOLOGY - Technical Guide for Solid Dispersion Development
PARTICLE DESIGN TECHNOLOGY – INTRODUCTION
The number of pipeline drug candidates that are poorly soluble, falling into Developability Classification System (DCS) Class IIa, IIb, and IV, continues to increase. The poorly soluble nature of these compounds leads to poor absorption, poor bioavailability, and increased pharmacokinetic variability. Clinically, these features result in drugs with poor efficacy and safety profiles. Because many of these drugs exist as crystalline materials, the answer may lie in creating an amorphous solid dispersion (ASD) for which spray drying technology is a key part of the manufacturing process. There are many articles on the theory and scientific principles underpinning the benefits of ASD. This article aims to introduce the reader to the steps involved in the development and manufacturing of an ASD via the spray drying process.
HOW DOES SPRAY DRYING WORK?
An ASD comprises a mixture of the API in its amorphous state, with at least one polymeric carrier excipient that has a single glass transition temperature, and no detectable crystalline structure. These mixtures are created by dissolving the API and the polymer excipient (or excipients) in at least one strong, volatile solvent, and then rapidly removing those solvents. The spray and evaporation rates are crucial: the evaporation must take place before either crystallization or phase separation has time to occur.
The high speed of the spray drying process makes it an essential tool in the creation of amorphous forms. The API is in solution, and the spray dryer very rapidly evaporates the solvent into a vapor, leaving behind the API as a dry solid. To achieve this, the solution is sprayed into a drying chamber through a narrow nozzle that creates very fine droplets, and solids form as the solvent evaporates. Spray drying of only the API is not sufficient as the particle will have a high tendency to rapidly re-crystallize. Therefore, a hydrophilic polymer is added to inhibit or retard recrystallization, as well as improve the solubility and dissolution rate of the API (Figure 1).
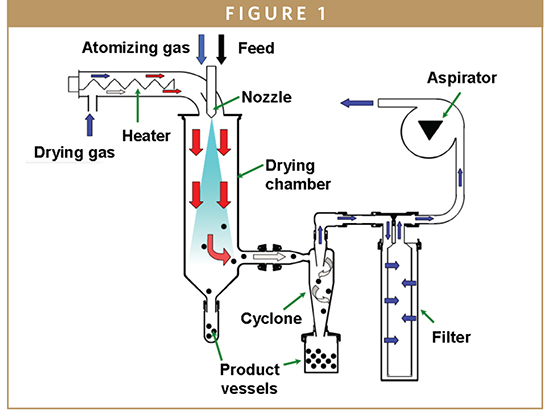
Particle size, density, compressibility, and residual solvent levels are some of the main parameters to assess the quality of the spray-dried particles and their robustness to be further incorporated into a final dosage form. The resulting particle size that is formed is important both in terms of performance and powder handling, and is determined by the spray pattern of the droplets of solution that emerge from the nozzle, as well as the tension and viscosity of the solution, the solids content, the spray rate, and the atomization gas that is used. Density and compressibility (important parameters in the ability of the powder to be formulated into a tablet) are related to particle size and the inlet and outlet temperature. The optimization of the inlet and outlet temperature range is critical for controlling the drying kinetics, which impact the final particle morphology. Any residual solvent will have an impact on physical stability.
BENEFITS OF CREATING ASDS
An amorphous dosage form is typically not the first choice for a drug formulator, as other approaches such as salt forms, co-crystals, and micronization, are likely to be less costly and require a lower degree of technical expertise to manufacture at larger scale. If none of these approaches give the desired solubility, then it is worthwhile to assess the development of an amorphous dosage form.
Cases in which this has proved to be the best option include APIs in which polymorphic form (different crystalline structures) control has been problematic, as the amorphous form gives process control over polymorphs. It can also assist where residual solvents or particle size distribution have been difficult to control during API synthesis.
The method also offers an alternative method when looking to increase the solubility in the gastric and intestinal fluids of APIs that fall into Class II of the Biopharmaceutical Classification System (BCS), or Class IIb (solubility-limited absorption) of the DCS. Amorphous forms are intrinsically more soluble, as there is no crystalline lattice energy to be overcome. It is advantageous to have a resultant finely divided powder that can be processed into any dosage form that can incorporate powders, including capsules and tablets.
Conversely, there are some APIs that are never likely to be appropriate candidates for an amorphous dosage form because of the high energy of the powders. These include APIs that are inherently unstable, where chemical degradation is possible, or where the API is particularly hygroscopic.
Manufacturing considerations can also remove the amorphous form as an option. Complex enhancement steps are required in the process, such as spray drying, hot melt extrusion (HME) or Wurster coating, and secondary processing, such as tray drying, granulation, or compression, increases the complexity of manufacturing. It should also be noted that there are usually more in-process, release, and stability testing requirements to be met for an amorphous dose than alternative methods.
PREFORMULATION SCREENING
The first step in creating the formulation is to screen a range of parameters that describe the API’s physicochemical characteristics and determine the composition of the formulation. These include the form in which the drug will be incorporated into the dose, such as a salt or the free form, and determining whether there are any known polymorphic forms or solvates whose formation will need to be monitored. The API’s baseline solubility at 37°C in different biologically relevant media [fasted-state simulated gastric fluid (FaSSGF), fasted-state simulated intestinal fluid (FaSSIF) and fed-state simulated intestinal fluid (FeSSIF)] should be established, along with its DCS categorization and the solubility limited absorbable dose (SLAD).
Additionally, the API’s solubility and stability in common volatile solvents (water, ethanol, methanol, isopropanol, hexane, acetone, dichloromethane, tetrahydrofuran, and ethyl acetate) should be assessed. The solubility of the most stable polymorphic form of the free-form API should be measured, and the stability study should be monitored at both ambient and accelerated temperatures for 3 to 7 days.
It is not uncommon that the API will not have sufficient solubility in a single solvent, so a combination of solvents can be used. In solvent combination screening, two to four solvents are selected that give the best solubility and stability results in the initial screen, and tests should be undertaken to further measure the solubility and stability of the API in 20:80 and 80:20 mixtures of all possible pairs of the solvents. The combinations that give the most acceptable solubility and chemical stability characteristics should then be used for those spray drying tests, but it is preferable that one predominate solvent is used whenever possible.
If multiple solvent systems with solubility greater than 50 mg/g and stability for 3 days at accelerated temperature are identified, then it is safe to move on to initial spray drying tests. If not, then solubility and stability need to be optimized, which can usually be achieved through solvent combination screening.
POLYMER SCREENING
Amorphous APIs by definition are in a high-energy, unstable state. The driving force for the API is to revert into its low-energy, stable crystalline state. Excipients, specifically polymers, are used to inhibit or delay crystallization, which in turn affects the physical stability, dissolution rate, supersaturation level, and bioavailability of the API.
Identifying the best polymer excipient, or a combination of them, is an important step in creating the optimal spray dried powder. Ideally, between two and six such polymers will be selected for screening; the four most common are hydroxypropyl methylcellulose (HPMC), polyvinylpyrrolidone (PVP), polyvinylpyrrolidone/vinyl acetate (PVP-VA), and hydroxypropyl methylcellulose acetate succinate (HPMCAS). Batches of between 1 and 5 g should be prepared with a 15% to 25% API loading in each polymer system – the same percentage loading should be used for each different polymer.
Once the test batches have been spray dried, the homogeneity of the amorphous product is assessed by differential scanning calorimetry (DSC). It is also important to assess the amorphous system’s potential to reach supersaturation levels in relevant biological media, such as simulated gastric fluid (SGF), FaSSIF, and FeSSIF.
After establishing the initial screening results, the next step is to focus on the optimal polymer choice and the best loading rate. Between one and three polymers that have appeared promising in the rapid screen are chosen, and batches of several loads ranging from 10% to 75% of active, based on pre-formulation data are produced. The polymers are sprayed at selective loads on a 1 to 5 g scale, and then kinetic solubility testing versus load is performed.
Physical and chemical stability of these samples is assessed in several ways. Samples at the various loading levels should be stressed to induce crystallization, if possible, and to allow for any chemical incompatibilities to be detected. In addition to DSC studies, powder X-ray diffraction (PXRD) experiments are performed to assess crystallinity, and high-performance liquid chromatography (HPLC) is used to assess chemical stability. Both water content and visual appearance are also assessed.
With these data in hand, the next step is to select a lead dispersion. Chemically incompatible polymers or loads can be pinpointed by comparing sealed and open dishes that have been left open to the atmosphere for a period of time, and if any degradation is observed, whether this is a result of the polymer or the exposure to moisture in the atmosphere. It may be possible to improve stability with appropriate packaging or moisture protection measures, and if this is the case, poor results at this stage will not necessarily result in failure of the dispersion.
Similarly, those polymers and loads that are physically incompatible should be identified. Phase separation or crystallization will be problematic if they occur in the finished dosage form, but again, it may be possible to avoid these with suitable packaging.
Those batches that do not fail for stability reasons can then be assessed for overall suitability and the selected samples screened individually, using a bio-relevant kinetic solubility test to identify which have the highest levels of solubility. The DCS is then revisited, to assess whether the spray-dried samples are likely to have improved properties if an amorphous dosage form is created. Ideally, the solubility of the final spray-dried system will be high enough to increase the DCS IIb rank to either DCS IIa or DCS I rank.
LABORATORY-SCALE SPRAY DRYING TESTS
The first step of spray dry process development is the running of feasibility batches, which can be prepared on a gram scale and are designed to identify the optimal solvent systems, polymer excipients, solids content and active loading level, as well as verifying the amorphous content and stability of the spray-dried powder.
There are broadly two designs of spray dryer that can be employed: the open loop system and the closed loop system. Both have their advantages and drawbacks; with an open loop system, the solvent saturation will be low; however, there is a much higher explosion risk because of the inevitable combination of oxygen, heat, and fuel. In contrast, the closed loop system does not pose this risk of explosion as the system is flooded with nitrogen rather than being open to the air. As well as being safer, it is also faster, more scalable, and the design is more environmentally friendly. On the down side, the solvent saturation is much higher.
Several small-scale spray dryers are available from commercial sources, and again, all have their advantages and disadvantages so, ultimately, the choice comes down to the nature of the material that is being spray dried. The ideal small-scale spray dryer will offer the ability to carry out particle engineering experiments that are representative of what will happen on a larger scale, and the ability to tailor particles for inhalation applications. It should give a high yield for platforms screening batches, be easy to customize, and offer continuous spraying.
SPRAY DRYING SCALE-UP
To move from the laboratory scale to commercial manufacture, the spray drying process must clearly be carried out on a much larger scale. The solvent system, polymer, and drug loading will have already been determined, and larger batches will need to be made for preclinical animal studies, GLP studies, and eventually GMP manufacturing to support clinical trials and commercial launch.
The sort of lab-scale spray drying equipment used for those small test batches is wholly insufficient in capacity to support larger scale work, as the maximum batch size is around one kilogram.
Spray dryers, such as a GEA Niro Mobile MinorTM machine, are used for the next step of development at pilot scale. This equipment permits continuous operation in a closed cycle system and has a maximum evaporative capacity of about 2 kg of water per hour, and the option of 10- to 350-L jacketed mixing systems.
There are two options for nozzle selection: a two-fluid nozzle or a high pressure nozzle. The former requires a peristaltic pump and atomization gas, and 0.5-1.5 mm tips are available. Particle size control can be achieved by altering the ratio of atomization gas to liquid flow rate: increasing the liquid flow rate will give larger particles; increasing the gas flow rate will give smaller particles.
With a high pressure nozzle, there is not atomization gas; rather, it employs a pressurized diaphragm pump. Tips range from 0.1 to 0.4 mm, and the spray angle can be varied from 45° to 120°. Increasing the flow rate increases the pressure, which gives a tighter distribution of smaller particles. In contrast to the two-fluid nozzle, which is only really appropriate at lab scale, the high-pressure nozzle can be used to make much larger quantities. In addition, where the two-fluid nozzle has very high consumption of nitrogen, none is needed for the high pressure nozzle.
When planning a scale-up, several parameters need to be considered in addition to the nozzle and tip size. These include pressure and liquid flow rate, the nature of the drying gas and its flow rate and temperature, and the outlet temperature in the control of residual solvent and saturation.
To remove residual solvent and reduce its impact on the stability of the final API, when spray drying on large-scale equipment, a secondary drying process is necessary, which will typically be carried out in a vacuum oven or granulation dryer depending on the scale. It is important that the spray-dried substance meets International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines for residual solvent Q3C(R6) and USP <467> residual solvents.
Drying studies should be run to check whether the solvent removal is adequate. This can be done by storing the material in a drying oven, sampling at multiple time points, and sampling for residual solvent via gas chromatography.
Roller compaction is typically performed as a final step to increase the particle size and density, and in this process, the powder is forced through two counter-rotating rollers to increase the density and compact the powder into ribbons. These ribbons are then milled to give uniform granules. The powder that emerges from the roller compactor has greatly improved flow properties, with a typical bulk density of 0.5 to 0.7 g per cubic centimeter. This is important when compressing tablets or manufacturing capsules, as it will give better weight and content uniformity to the final dose form.
DOSAGE FORM SELECTION
With a dispersion in hand that is both physically and chemically stable, as well as providing adequate solubility enhancement, the next step is to look at using it to create the most appropriate solid dosage form, whether this is a tablet, a capsule, or some alternative format. It is important that potential excipients required to make the dosage form should be selected and tested for compatibility with the spray-dried powder; however, the manufacturability and disintegration must also be considered when selecting excipients and developing a formulation.
When assessing the compatibility of excipients, both binary mixtures and design of experiment (DoE) studies are suitable. Previous stability data gathered with the crystalline drug should be reconsidered, and all samples should be made using the lead amorphous dispersion material. The use of the final amorphous intermediate is crucial for excipient compatibility studies, as amorphous forms are more sensitive to degradation, but it is also important to consider moisture protection in the study.
Formulation development efforts should be focused on creating dosage forms with fast disintegration times, with dissolution being monitored for gelling. It may be appropriate to consider more aggressive disintegration approaches if initial attempts produce long disintegration times, or slow dissolution rates. These might include the incorporation of high levels of bicarbonate, an osmogen (high-water solubility excipients), or a super-disintegrant. All in vitro formulation development work should be confirmed with in vivo studies whenever possible and the neat dispersion suspended in water along with the formulated dispersion product should be compared in a relevant model to ensure poor disintegration is not leading to lower than expected exposure levels. If the final dosage form is not formulated so that rapid, consistent disintegration is achieved, then the increased solubility can be quickly negated by the poor release of the spray dried intermediate. Formulation processes to increase density and reduce surface area can slow down the spray-dried intermediate’s wetting rate, allowing the final dosage form to disintegrate fully without forming a gel.
The quick identification of a suitable dispersion is not a trivial exercise, not least because disintegration, flow, and density challenges are amplified when developing a spray dried amorphous product. Although pursuing ASD formulation may extend timelines and introduce complexities not seen with traditional tablets and powder-filled capsules, implementation of DoE and working with an experienced partner can help avoid bottlenecks and program delays. Time and cost savings can be realized by formulating DCS IIb drugs to address bioavailability challenges prior to first-in-human studies and avoid the need to go back and re-formulate. A study at GlaxoSmithKline showed that for DCS IIb compounds, it takes 2 additional years of performing clinical studies to reach a decision to stop development because a compound does not have adequate exposure to reach sufficient efficacy.1 Spray drying and amorphous formulations can be very powerful tools to increase solubility and bioavailability within a traditional dosage forms such as a capsule or tablet.
REFERENCE
1. Hann MM, Keserü GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discov. 2012;30;11(5):355-65. doi: 10.1038/nrd3701.
To view this issue and all back issues online, please visit www.drug-dev.com.

Nathan Barksdale is a Scientist at Catalent Pharmatek SD and is an expert on early stage development technologies for preclinical through Phase II dosage form development and cGMP manufacturing. He has over 10 years of experience in the pharmaceutical development and manufacturing industry working with small molecules and peptides and has an extensive background in preclinical through clinical drug development and cGMP manufacturing. He possesses a strong knowledge base in oral formulation development and manufacturing, complex process trains, and highly potent and cytotoxic compounds. Mr. Barksdale earned his BS in Pharmacological Chemistry from the University of California, San Diego.

Elizabeth Hickman is Strategic Marketing Director, Drug Delivery Solutions at Catalent Pharmatek SD. Previously, she was Director of Sales and Marketing at Pharmatek prior to its acquisition by Catalent. Prior to joining Pharmatek, she held marketing and sales management positions with increasing responsibility at Pharmaceutics International Inc. (Pii), PharmaForm, and UCSD, where she developed an extensive understanding of marketing, management, and business development. She brings more than 10 years of contract development and manufacturing industry experience to Pharmatek. She instills a consultative approach to the development of new business opportunities, while driving sales through data-driven marketing programs. Ms. Hickman earned her MBA from San Diego State University and her BA in Microbiology from the University of Texas in Austin.
Total Page Views: 6195



