


Issue:October 2015
ORODISPERSIBLE TABLETS - Advanced Drug Delivery for Repositioning Promethazine for Improved Application & Patient Outcomes
INTRODUCTION
Promethazine is a derivative of phenothiazine that blocks histamine H1 receptors.1 It is commonly used to treat a variety of allergies and allergic reactions.2 This medicine is also used to control dizziness, motion sickness, nausea, and vomiting.3-5 The dosage of promethazine comes in three forms: syrup, tablet, or elixir. Usually, 25 mg of promethazine is taken orally every 4 to 6 hours when used to treat nausea and vomiting.2 The delivery of promethazine via digestion can be problematic for someone who has undergone chemotherapy, anesthesia, or surgery due to nausea and emesis. Therefore, it is common for nauseated patients to take promethazine via direct intravenous injection or suppository. These inconvenient delivery methods can be avoided by the production of quick-release, orodispersible promethazine tablets.
Drug delivery by absorption through oral mucosa offers advantages such as direct absorption into the systemic circulation, thus avoiding presystemic elimination via the gastrointestinal tract.6 The sublingual mucosa is relatively permeable due to its thin membrane and large veins, allowing quick absorption and bioavailability of drugs due to high blood flow. However, sublingual delivery is not appropriate for sustained delivery formulations.7 When delivering orodispersible promethazine tablets, the sublingual mucosa is a well-suited area for fast release, bringing rapid bioavailability of the drug.8-11
To overcome the aforementioned delivery challenges, a formulation of orodisperisble tablets (ODT) at three different compression settings (low, medium, and high) have been designed and characterized for friability, hardness, wetting time, disintegration, and in vitro dissolution. In order to determine the effective formulation and compression setting for manufacturing of rapid release of ODT promethazine tablets, these parameters were statistically analyzed.
MATERIALS
Promethazine and crossprovidone were obtained from Spectrum Chemical Manufacturing Corporation, USA, and ISP Technologies, USA, respectively. Lactose monohydrate (316 Fast Flo) was provided by Foremost Farms, USA. Mannitol (Pearlitol) 300DC was provided by Roquette Corporate, USA. Camphor was obtained from Acros Organics, USA. Sodium Stearyl Fumarate, was purchased from JRS Pharma, Germany.
METHODS
Formulation Study Design
A simplex lattice design was used in order to develop response surface and contour plots.12 The amount of lactose (A), camphor (B), and mannitol (C) were selected as the independent variables. The three variables were tested at three different levels – low, medium, and high-compression settings. Disintegration time (Y1), percent release at 30 seconds (Y2), and percent release at 1 minute (Y3) were all used as dependent variables.
Preparation of Tablets
Tablets were made using a direct blend and compression method with ¼-inch standard concave tooling. For each formulation, the tablets were made using three different compression forces – low, medium, and high of ~5 kN, ~10 kN, and ~20 kN, respectively. The batches were then characterized, according to USP standards for variability of the tablet dimensions and weight, friability, hardness, disintegration, and dissolution times.13,14
Friability
Friability was tested by weighing each tablet, then setting the Friability Tester Model FTV-2 (Dr. Schleuniger Pharmatron, Switzerland) at 50 revolutions per minute for 1 minute. The largest piece of the remaining tablet was weighed to determine percent loss.
Hardness
Each tablet was aligned in the center of the VK 200 Tablet Hardness Tester (Varian, USA), and the hardness was recorded in kiloponds (kp).
Wetting Time
Wetting time was studied using a published method.15 In brief, a crude filter paper of 10-cm diameter was placed on the bottom of a 10-cm diameter Petri dish, and 10 mL of water containing methylene blue was added. Each tablet was carefully placed in the petri dish, and the wetting time was recorded when the water had reached the upper surface of the tablet.
Disintegration
Disintegration time was measured in sink conditions using de-ionized water in DT2-IS Disintegration Tester (Dr. Schleuniger Pharmatron, Switzerland). When all the granules passed through the basket, the disintegration time (seconds) was recorded.
Dissolution
Release of the drug was studied for each batch using Vision Classic 6 (Hanson, USA) USP Dissolution Apparatus II in 900 mL of phosphate buffer release medium pH 6.8 at 37±0.5ºC with paddle speed at 100 rpm.16 3-mL samples were collected at predetermined time points (0, 0.5, 1.0, 2.0, 3.0, 5.0, and 10.0 mins) and replaced with 3 mL of medium. The sample was filtered using a coarse filter paper to remove any particulates, cooled at room temperature, and promethazine content determined by absorbance at 253 nm.
Statistical Analysis
Simplex lattice matrix was utilized for analysis of three formulation variables at three levels and their effect on dependant variables. All data was obtained as a mean of at least three experiments ± standard error. The differences in effect on dependant variables of the formulations were studied by two-way ANOVA using DesignExpert 8.0 software to obtain regression fit and surface response graph. 5% significance level (p value ≤ 0.05) was utilized for all statistical analyses.
RESULTS
In order to understand the effect of excipients on the properties of the tablets, six various formulations were designed for testing tablets containing various amounts of lactose, camphor, and mannitol. The effect of the formulation composition has been studied by assessment of physical parameters of the tablets – friability, hardness, wetting time, and disintegration time.
All tablets demonstrated friability of less than 1% loss and are in accordance with USP regulations of ODT tablets, and therefore, can endure shipping and handling without breaking.17 The hardness of the formulations at low, medium, and high compression settings were in the range of 3.03 ± 0.19 to 4.17 ± 0.03 (kP), 4.03 ± 0.17 to 6.27 ± 0.13 (kP), and 4.73 ± 0.23 to 8.10 ± 0.5 (kP), respectively.
The hardness for each ODT formulation increased with increasing compression setting. However, ODT 2 showed only a slight difference between medium (5.53 ± 0.15 kP) and high (5.53 ± 0.27 kP) compression tablets. The steepest increase in hardness was displayed with ODT 1, and the most gradual increase was seen with ODT 4.
Tablets at low, medium, and high compression were tested and found to have wetting times in the range of 9.53 ± 0.99 to 49.30 ± 24.05 (secs), 9.93 ± 2.02 to 49.40 ± 6.78 (secs), and 26.30 ± 3.01 to 41.40 ± 9.75 (secs), respectively; disintegration times in the range of 8 ± 0 to 163.67 ± 16.13 (secs), 18.67 ± 3.67 to 198.33 ± 13.64 (secs), and 95.33 ± 2.91 to 303.33 ± 29.67 (secs), respectively. Dissolution times varied with compression setting and formulation (Figure 1), but ODT 1 demonstrated the fastest release at the low compression setting. Overall, all tablets resulted in rapid disintegration, making them suitable for consideration for orodispersable delivery.
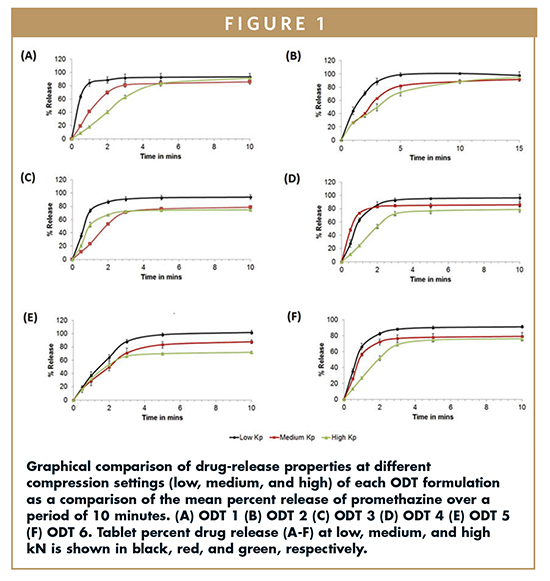
In order to understand the effect of the formulation variables (A, B, and C, Figure 1) on the release of the drug from the tablet, the dissolution was studied in vitro. This study showed a general trend in that the low kN tablets released first, followed by the medium kN tablets, and high kN tablets released last.
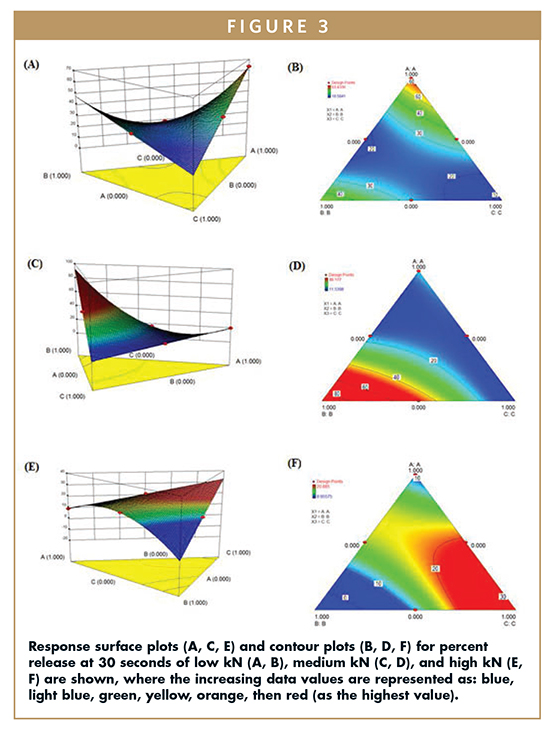
In order to systematically analyze the camphor, and mannitol) on tablet properties, a simplex lattice matrix was used. The coded values (A, B, and C represent lactose, camphor, and manitol, respectively) were used in each design. Each design was separately imputed into the SAS to analyze the effect of lactose, camphor, and mannitol on disintegration time (Figure 2), drug release at 30 seconds (Figure 3), and drug release at 1 minute.
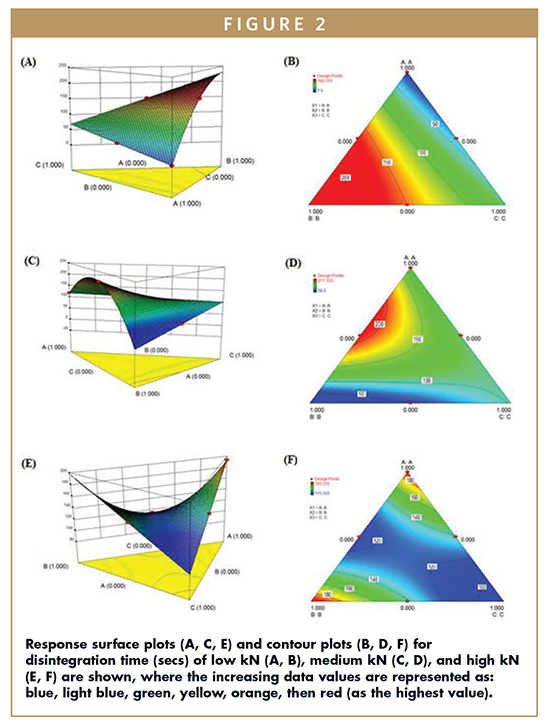
To ensure drug is successfully delivered via orodispersable tablet, rapid disintegration is essential. Equations 1-3 demonstrate the importance of camphor (B) in rapid dissolution that can be described by a negative coefficient effectively decreasing the time for the tablet to disintegrate (-33854.56, -11324.53, +11267.94 for low, medium, and high kN, respectively). The effect of camphor decreases with compression as evidenced by a more positive coefficient for disintegration time equations for low, medium, and high kN (Equations 1-3). Lactose (A) increased disintegration time of the tablets, and this effect is more pronounced with increased compression setting as the coefficients were +7.50, +118.83, and +193.33 for low, medium, and high kN, respectively. Mannitol had a negative effect on disintegration time at low kN, resulting in an increase of disintegration time. However, at high kN, mannitol had a positive effect on disintegration with +34153.56, +11425.53, and -10985.27 constants at low, medium, and high kN, respectively.
Equations 1-3: Disintegration Time
1. Low kN: Y1 = + 7.50A – 33854.56B + 34153.56C + 68367.45AB -68170.78AC
2. Medium kN: Y1 = + 118.83A – 11324.53B + 11425.53C + 23256.74AB – 22647.40AC
3. High kN: Y1 = + 193.33A – 11267.94B + 10985.27C – 22461AB + 22139.21AC
The effect of the independent variables A, B, C (lactose, camphor, mannitol) on drug release were in line with disintegration time. In this study, percent release of promethazine at 30-seconds and 60-seconds time points were considered (Equations 4-9). Lactose had a positive effect on drug release regardless of the compression setting (low, med, and high kN) with coefficients +63.43, +19.47, +8.96 (30 secs, Equations 4-6) and +84.21, +41.17, +18.41 (60 secs, Equations 7-9). This effect appears to decrease with increase in compression. Camphor (independent variable B) positively affected the release with increasingly negative impact with increase in compression – coefficients of +7418.57, +3173.61, -2825.64 (30 secs), and +7706.38, +12953, -8542.79 (60 secs) for low, medium, and high kN, respectively (Equation 4-9). Mannitol (independent variable C) had a negative effect on drug release, which became less pronounced, and even positive at higher compression with coefficients of -7363.42, -3077.26, +2848.08 (30 secs) and -7580.50, -12806.50, +8591.98 (60 secs) for low, medium, and high kN. In other words, mannitol demonstrates more of a binding property with lower compression and is more contributive to disintegration at higher kN.
Equations 4-6: Percent Release at 30 Seconds
4. Low kN: Y2 = + 63.43A + 7418.57B – 7363.42C – 14889.68AB + 14742.14AC
5. Medium kN: Y2 = +19.47A + 3173.61B – 3077.26C – 6320.46AB + 6161.74AC
6. High kN: Y2 = +8.96A – 2825.64B + 2848.08C + 5699.20AB – 5630.53AC
Equations 7-8: Percent Release at 60 Seconds
7. Low kN: Y3 = + 84.21A + 7706.38B – 7580.50C – 15433.93AB + 15286.10AC
8. Medium kN: Y3 = + 41.17A + 12953.61B – 12806.50C – 25873.03AB + 25264.39AC
9. High kN: Y3 = +18.41A – 8542.78B + 8591.98C + 17177.87AB – 17012
The effect of combining all of the formulation components were computed, and the data for ODT 1 shows highest rapid disintegration with highest percentage of drug release at 30 sec and 60 sec. Interestingly, compression of the tablet also plays role, where ODT 4 showed the shortest disintegration time with highest percentage of drug release as compared to other formulations at medium compression setting. At the highest compression settings, ODT 5 had the most rapid disintegration time, and ODT 3 showed the most rapid release. This may be due to increased formation of tight granules in the tablet at high compression, resulting in prolonged and more variable release.
DISCUSSION
The optimal characteristics of fast-release, orodispersible promethazine tablets in this study are described as: weight, diameter, and thickness and were within USP range below 5% RSD with appropriate tablet friability of less than 1% loss for all of the formulations. With hardness ranging from 4 to 5 kP, the tablets are expected to withstand the normal forces experienced during handling and transport. Short wetting and disintegration times, and at least 90% drug release within 5 minutes to allow fast disintegration in the mouth, promote adequate release of promethazine and time for absorption through the buccal mucosa. Disintegration time for the medium kN tablets was 4 to 18 seconds and higher for batches with higher punching pressure ranging between 1 to 4 minutes. The wetting time of the low and medium compression tablets show a trend in which the concentration of camphor has a positive impact, decreasing wetting time, whereas the concentration of mannitol has a negative impact on wetting time.
The response surface and contour plots for disintegration time and drug release demonstrate overall trends with respect to the independent variables. With a pronounced peak at independent variable B, camphor has a negative impact on the disintegration time of low, medium, and high kN tablets (Figure 2). The response surface plot for medium compression tablets (Figure 2C) has a saddle configuration in which mannitol has a positive impact on disintegration time as the concentration of camphor has the most positive impact on disintegration time, and the highest point is where the concentration of variable C (mannitol) is the lowest (Figure 2A & B). The response surface plot for high kN tablets shows a deep valley in which the lowest points are at the highest and at the lowest concentrations of mannitol, and the highest points are at the highest concentrations of lactose and camphor. This shows that at a high compression setting, the concentration of lactose and camphor both have a negative impact on disintegration time (Figure 2).
Considering the percent drug release at 30 seconds, mannitol has a negative impact on percent release of low compression tablets. In addition, both variable A (lactose) and variable B (camphor) have a positive impact on percent release, allowing more of the active ingredient to be released at 30 seconds; however, lactose shows the most positive impact. This analysis can be seen in Figure 3A & B. At a medium compression setting, camphor has a substantial positive impact on percent release at 30 seconds, shown by a peak in the response surface plot (Figure 3C). Also, mannitol has a negative impact on percent release of medium kN tablets, seen as the lowest point in Figure 3C. At high compression setting, camphor becomes less important for rapid drug release due to impact drug release, whereas mannitol has a positive impact on percent release at 30 seconds. This is represented in Figure 3E & F. Drug release at 60 seconds shows identical trends at the distinct compression settings as the percent release at 30.
In this study, the formulations for delivery of promethazine have been successfully prepared and characterized. The importance of the punching pressure during the preparation process is considered as the contribution of the independent variables to the tablet properties may vary. Out of the six formulations considered in this study, ODT 4, made with a high punching pressure, is the most suitable candidate for orodispersible promethazine delivery with hardness of 4.73 ± 0.23 and friability of 0.39 ± 0.14% – both parameters are compliant with USP regulations, and a wetting time of 20.37 ± 4.97 seconds, allowing quick disintegration in the mouth. Most importantly, in vitro dissolution testing demonstrated rapid and sustained drug release of 90% over a 3-minute timespan.
CONFLICT OF INTEREST
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
ACKNOWLEDGMENTS
The authors wish to thank CoreRx, Inc. research and manufacturing departments for assisting with tablet manufacturing. We acknowledge the funding received from Florida High Tech Corridor.
REFERENCES
1. Suzuki A, Yasui-furukori N, Mihara K, et al. Histamine H1-receptor antagonists, promethazine and homochlorcyclizine, increase the steady-state plasma concentrations of haloperidol and reduced haloperidol. Ther Drug Monit. 2003;25(2):192-6.
2. Promethazine Drug Info: http://www.nlm.nih.gov/medlineplus/druginfo/meds/a682284.html.
3. Kolahian S, Jarolmasjed SH. Antiemetic efficacy of promethazine on xylazine-induced emesis in cats. Can Vet J. 2012;53(2):193-5.
4. Zur E. Nausea and vomiting in pregnancy: a review of the pathology and compounding opportunities. Int J Pharm Compd. 2013;17(2):113-23.
5. Fahler J, Wall GC, Leman BI. Gastroparesis-associated refractory nausea treated with aprepitant. Ann Pharmacother. 2012;46(12):e38.
6. Szabó B, Hetényi G, Majoros K, Miszori V, Kállai N, Zelkó R. [Formulation aspects and ex-vivo examination of buccal drug delivery systems]. Acta Pharm Hung. 2011;81(4):165-72.
7. Harris D, Robinson JR. Drug delivery via the mucous membranes of the oral cavity. J Pharm Sci. 1992;81(1):1-10.
8. Ikeuchi-takahashi Y, Sasatsu M, Onishi H. Evaluation of matrix type mucoadhesive tablets containing indomethacin for buccal application. Int J Pharm. 2013;453(2):454-61.
9. Sakata O, Onishi H. In vivo absorption study of ritodrine hydrochloride in the buccal administration to rats. Drug Dev Ind Pharm. 2013;39(9):1414-20.
10. Choi SG, Baek EJ, Davaa E, et al. Topical treatment of the buccal mucosa and wounded skin in rats with a triamcinolone acetonide-loaded hydrogel prepared using an electron beam. Int J Pharm. 2013;447(1-2):102-8.
11. Gayrard V, Lacroix MZ, Collet SH, et al. High bioavailability of bisphenol A from sublingual exposure. Environ Health Perspect. 2013;121(8):951-6.
12. Patel DM, Patel NM, Pandya NN, Jogani PD. Gastroretentive drug delivery system of carbamazepine: formulation optimization using simplex lattice design: a technical note. AAPS PharmSciTech. 2007;8(1):11.
13. USP 2012. Dissolution standard <711>: https://mc.usp.org/sites/default/files/documents/GeneralChapterPDFs/c711_USP36.pdf .
14. Abed KK, Hussein AA, Ghareeb MM, Abdulrasool AA. Formulation and optimization of orodispersible tablets of diazepam. AAPS PharmSciTech. 2010;11(1):356-61.
15. Aburahma MH, El-laithy HM, Hamza Yel-S. Preparation and in vitro/in vivo characterization of porous sublingual tablets containing ternary kneaded solid system of vinpocetine with îcyclodextrin and hydroxy Acid. Sci Pharm. 2010;78(2):363-79.
16. Aburahma MH, El-laithy HM, Hamza Yel-S. Preparation and in vitro/in vivo characterization of porous sublingual tablets containing ternary kneaded solid system of vinpocetine with îcyclodextrin and hydroxy Acid. Sci Pharm. 2010;78(2):363-79.
17. Meher, DA. Formulation and development of fast-disintegrating naproxen tablets using simplex lattice design. Journal of Chemical and Pharmaceutical Research, 2011; 3(6):521-526.
To view this issue and all back issues online, please visit www.drug-dev.com.
 Dr. Srinvias M. Tipparjau is faculty at University of South Florida College of Pharmacy working as Assistant Professor in the area of cardiovascular diseases, drug development, and innovation. Currently, he is focused on understanding the mechanisms for diabetes, arrhythmias, and heart failure. He directs research efforts and established a research team and funding for projects including drug development and formulation design using innovative technologies.
Dr. Srinvias M. Tipparjau is faculty at University of South Florida College of Pharmacy working as Assistant Professor in the area of cardiovascular diseases, drug development, and innovation. Currently, he is focused on understanding the mechanisms for diabetes, arrhythmias, and heart failure. He directs research efforts and established a research team and funding for projects including drug development and formulation design using innovative technologies.
Anastasia Groshev is currently a medical doctorate candidate at the University of South Florida Morsani College of Medicine. Her work in research focuses on development of drug delivery forms for penetration of blood barriers and sustained release for treatment of brain tumors, Alzheimer’s disease, and glaucoma.
Danielle Dantuma is a Research Assistant at the University of South Florida College of Pharmacy. She is Operations Coordinator at the BRIDGE Pharmacy Clinic, Secretary of International Health Service Collaborative, and CIC Program Coordinator of the Student National Pharmaceutical Association chapter at USF.
Brian McMillan is the Vice President of Product Development and Co-founder of CoreRx, Inc. in Clearwater FL, and Adjunct Assistant Professor of Pharmacy at The University of South Florida.
Dr. Todd R. Daviau is the Founder, President, and CEO of CoreRx, Inc., bringing 27+ years of pharmaceutical experience and knowledge to the project. Prior to CoreRx, he served as VP of the Analytical and Pre-Formulation Laboratory for MDS Pharma Services and Strategic Analytical Team member with Guilford Pharmaceuticals, where he was involved in the development of Natrecor and Gliadel.
Matthew MacPherson is a graduate student at the University of South Florida, College of Engineering.
Dr. Yashwant Pathak is Associate Dean for Faculty Affairs at College of Pharmacy, University of South Florida in Tampa, FL. His areas of research include drug delivery systems and characterization in animal models.
Dr. Vijaykumar Sutariya is an Assistant Professor in the Department of Pharmaceutical Sciences at the University of South Florida. His areas of expertise are formulation development, controlled ocular drug delivery systems, and brain targeted drug delivery systems.
Total Page Views: 6108



