


Issue:October 2017
LIPID-BASED DELIVERY - Are Lipid-Based Drug Delivery Systems in Your Formulation Toolbox?
ABSTRACT
This article reviews the causes of poor bioavailability for drugs. It provides an introduction to lipid-based drug delivery systems, and how the formulation approach can be used to overcome impediments to good bioavailability of therapeutic actives, including poor water solubility, low permeability, and degradation by stomach acid or enzymes in vivo.
INTRODUCTION
Oral bioavailability, simply defined, is a measure of the quantity of drug absorbed into the systemic circulation following oral administration of drug. A variety of physicochemical and or physiological mechanisms can negatively impact the rate and extent of a drug’s oral absorption. The unfavorable physical chemical properties of a drug may include poor aqueous solubility and low dissolution rate (frequently correlated to each other),1-6 an octanol-water partition coefficient below 17 and above 3 to 5,8 which translates into poor permeability through the gastrointestinal tract wall, and a chemical structure that renders the drug molecule prone to degradation via acid catalyzed reactions within the stomach and/or via enzymes including proteases and esterases within the gastrointestinal mucosa and blood stream.9-11 The physiological mechanisms that can decrease bioavailability of orally administered drugs include pre-systemic elimination by digestive enzymes, P-glycoprotein mediated efflux transport of drugs back into the gastrointestinal tract lumen,1,18-21 and/or first-pass metabolism (hepatic elimination) whereby a drug is absorbed in the intestine and is shunted to the liver via the portal circulatory system and immediately metabolized.1,12-17
In addition to chemical modifications to the drug molecule, there are multiple formulation approaches that can be used to overcome the aforementioned obstacles and improve the bioavailability of a poorly absorbed drug. These include alteration of solution pH and/or salt formation, enteric coating, solubilization by complexation, solid dispersions, and micronization and nanoization.1,22-49 The focus of this article is the use of lipid-based formulations to overcome barriers to bioavailability.
INTRODUCTION TO LIPID-BASED DOSAGE FORMS
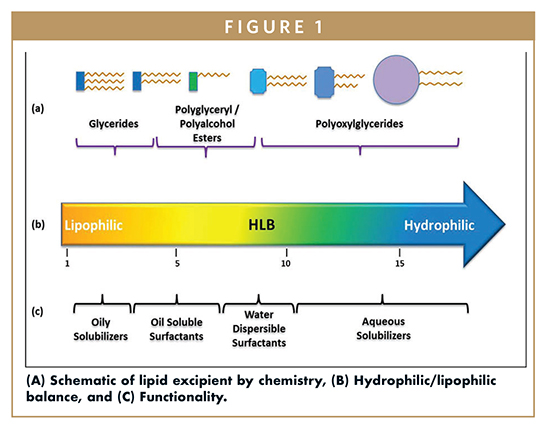
Lipid-based drug delivery systems (LBDDS) is a wide-ranging designation for formulations containing a dissolved or suspended drug in lipidic excipients. Lipids are esters of fatty acids – lipophilic hydrocarbon chains linked to a hydrophilic group like glycerol, polyglycerol, or polyalcohol (Figure 1). The melting range, solubilization capacity, and miscibility properties of the excipient are defined by the fatty acid chain length and degree of unsaturation. The amphiphilicity or dual polar and non-polar nature of lipids is characterized by the Hydrophilic Lipophilic Balance (HLB), a measure of the excipient dispersibility in aqueous media (Figure 2). Briefly, the functionality of the lipid excipients is connected to its chemistry (Figure 1).
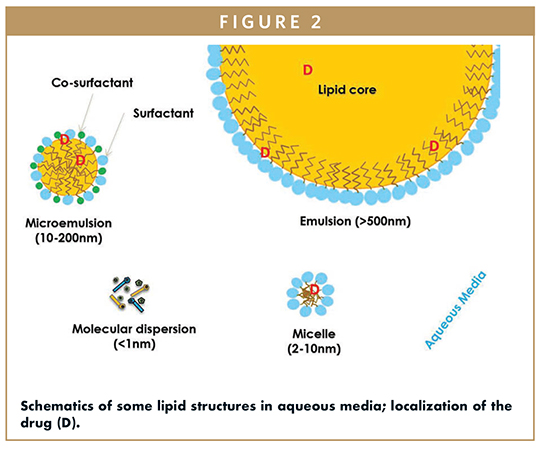
LBDDS can be as simple as a drug in oil to a more complex formulation that is designed to spontaneously emulsify upon contact with aqueous media. Such formulations are self-emulsifying drug delivery system (SEDDS) or self-microemulsifying drug delivery system (SMEDDS). Figure 3 helps visualize this spontaneous process, within seconds of introducing a small droplet of Labrafil® M 2125 CS in water, emulsification is observed. This excipient has a practical HLB of 9.

LBDDS can be used as drinking solutions, filled into hard or soft capsules, or incorporated into tablets; they maybe liquids, semi-solids, or solids at room temperature. As Savla and co-workers discussed, LBDDS provide an adaptable platform to deliver APIs that possess impediments to suitable bioavailability.50 The versatility of the platform originates from the multitude of excipients that are available to create formulations with targeted properties, including enhanced solubility and permeability, sustained release, etc. The large selection of excipients available to the formulator, in combination with how changes in composition affect performance (solubilization, permeation, and stability) of the formulation before and after digestion can make development of a LBDDS seem complex. Fortunately, lipid-based formulations have an extensive history of development and use to enable a systematic and fast approach for their development. Excipient suppliers, like Gattefossé, offer expert assistance to formulators in the form of guidelines, technical articles, and application laboratories and will work with formulation scientists to help solve their problems.
Owing to the versatility of LBDDS in solving drug delivery issues for drugs with high formulation barriers, there are many approved and marketed lipid-based drug products. Strickley reviewed these in 2007.51 Recently, 36 commercialized drugs consisting of LBDDS were reviewed.50 The excipients presented in these reviews have a well-known and long-term track record for safety, and have food or generally recognized as safe (GRAS) status.
PROBLEMS SOLVED BY LBDDS
Solubilization of Poorly Water-Soluble Drugs
Hauss has pointed out that over 70% of new drug candidates have low solubility values in water.4 Approximately 40% of lipophilic drug candidates that demonstrate good pharmacological activity do not reach market because low aqueous solubility compromises bioavailability and leads to poor pharmacokinetic performance and low exposure.1 The trend is likely to continue. No matter how promising the pharmacological activity of a drug, its inability to dissolve in the GI tract renders it ineffective. However, lipid-based dosage forms can be utilized to salvage good therapeutic agents that have low aqueous solubilities.
LBDDS provide the drug in a fully to partly solubilized state, and most importantly, maintain solution of the drug until it is absorbed. The drugs remain in the solubilized state because LBDDS self-emulsify (Figures 2 & 3), and/or form emulsions upon digestion. During digestion, oils in the LBDDS undergo lipolysis to form fatty acids and monoglycerides, which combine with components in the gastrointestinal fluids to form mixed micelles that can assist in maintaining the drug in solution.52
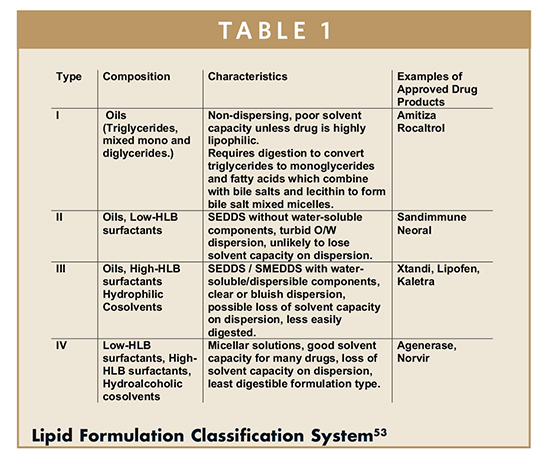
Lipid formulations are categorized inthe Lipid Formulation Classification System by their formulation components, hydrophobicity, dispersibility, and digestability (Table 1).53 A Type I formulation consists of triglycerides (oils), Type II adds water-insoluble and dispersible surfactants to the oils, Type III incorporates water-soluble surfactants and hydrophilic cosolvents (Transcutol®, ethanol, PEG), and Type IV contains no oils and is composed solely of surfactants and hydrophilic co-solvents. Type I formulations are non-water dispersible and rely on the body’s natural lipid digestion processes to form mixed micelles that act as a natural detergent for the drug. Figure 4 shows an emulsion formed through digestion of alpha-tocopherol quinone in a triglyceride. Type II formulations generally form course emulsions in water and are digested to form mixed micelles. Type III formulations form spontaneously as emulsions or micro/nanoemulsions upon dispersion in water, and may rely on some digestion to assist or maintain the drug in solution. Type IV formulations form micellar solutions upon dispersion in water and are least digestible. The common feature of Type I to Type IV formulations is solubilization through micellization in vivo. This is in contrast to other approaches, like solubilization by dissolving a drug in an organic co-solvent in which large reductions of solubility are experienced upon dilution. The formulator must understand that categorization is not predictive of in vivo performance, for example Type I formulations may perform best with highly lipophilic compounds of low melting point, but such compounds will not perform well if they are formulated with Type IV formulations. Hence the choice of formulation type will depend on the molecule being delivered.

Numerous examples exist in the literature that demonstrate the solubility and bioavailability enhancement achievable with LBDDS; a non-exhaustive list of active ingredients include progesterone, halofantrine, docetaxel, carvedilol, piroxicam, nifedipine, and curcumin.54-58
A potential advantage of LBDDS is that the drug is delivered in solution to the gastrointestinal tract, obviating the need for dissolution. The drug remains in solution through micellar solubilization brought about by digestion and/or self-emulsification. The challenge to the formulator is to discern which excipient(s) and what proportions of them will solubilize the dose.
Alskär and co-workers analyzed solubilities of molecules in various lipidic solvents and surfactants with the goal of establishing predictive solubility relationships.59 Some noteworthy findings from this work are: Lipophilic drugs with low melting point (< 150°C) are often soluble in oils or oily vehicles. The solubility of a drug in a mixture of lipidic excipients can be expressed as the sum of drug solubilities in the individual excipients, with each solubility value weighted by the excipient mass fraction in the formulation. Lastly, solubility values for a given molecule are similar in like solvents; for example, asolubility value in PEG400 can be used as an estimate for solubility values in other ethoxylated solvents like Transcutol®. This research provides a framework to estimate solubilities of molecules in lipidic excipients, and eases the work of the preformulator and formulator to select excipient(s) and their proportions to enable the targeted drug load.
Enhancement of Intestinal Permeability
To be absorbed into the systemic circulation, a drug molecule must pass through the GI wall. The resulting colloidal dispersions brought about by digestion of the LBDDS improve the drug affinity for the protective aqueous monolayer (or unstirred water layer) lining the lumen and facilitate the conditions for API permeability.60 This effect relates to excipients such as caprylocaproyl polyoxyglycerides (Labrasol®), which improves the drug transport through intestinal cell membranes and its effect on the opening of the epithelial tight junctions.61-65
Earlier studies, based on the in vitro cell-based model, pointed to membrane fluidity and efflux inhibition as other potential mechanisms driving intestinal permeability. Of late however, scientists are increasingly focusing on the supersaturation of API in the GI lumen as the leading mechanism for enhancing permeability. Recent studies suggest that initially, API supersaturation (an increase in free drug fraction) results from a decrease in the solubilization capacity of the LBF following dispersion in the gastric and intestinal fluids in the small intestine, creating pressure for API absorption.61,66-67 A subsequent imbalance created between the initial solubilized drug concentration in the GI fluids and drug solubility in the colloidal species formed post-dispersion and digestion is another factor contributing to API supersaturation. Meanwhile, the lipid metabolites are absorbed contributing to further reduction in the solubilization capacity of the remaining colloidal phases during digestion, thus promoting ongoing supersaturation.66-67
Protection From Enzymatic/Chemical Degradation
Hetényi and co-workers recently demonstrated that therapeutic peptides, leuprorelin, insulin, and desmopressin could be paired with sodium docusate and loaded into a SEDDS formulation.68 The researchers exposed the formulated peptides to intestinal proteases (α-chymotrypsin, trypsin, and elastase) and glutathione. They observed that no degradation of the peptides occurred in the SEDDS formulation, which was consistent with observation that the proteases and glutathione were ≤ 0.1% soluble in the oily SEDDS. This work strongly suggests that a LBDDS can protect sensitive peptide APIs from water-soluble reactants and degradation mechanisms that require an aqueous environment.
Reduction of First-Pass Metabolism
The triglycerides in a LBDDS are digested by the natural lipolysis process in the GI tract to form fatty acids and monoglycerides. Fatty acids can be absorbed by the hepatic and/or lymphatic route, and the distribution between the routes is dependent on the hydrocarbon chain length. Fatty acids with hydrocarbon chains below 12C tend to bind to albumin, which renders them water soluble. As a result, they passively diffuse through the epithelial cells lining the intestine and are taken up by the blood stream through the portal vein before being transferred to the liver.
Fatty acids with a chain length of 14C or longer, due to their hydrophobicity, can be substrates for transporting proteins into the cells, where they can be resynthesized into lipoproteins (known as chylomicrons) for uptake by the lymphatic route.
The unsaturated long-chain fatty acids (LCFA), in particular, are known to stimulate chylomicron secretion and increase the lymphatic uptake. They have been shown to enhance the bioavailability of certain drugs, such as saquinavir, ontazolast, halofantrine, by preferential absorption through the lymphatic transport system and consequently decreased first-pass metabolism of the API in the liver.69-71
Because absorption by the lymph means bypassing the liver, the co-formulation with LCFAs can be a promising strategy for drug actives that are extensively metabolized in the liver. Enhanced lymphatic absorption is important in oral delivery primarily for highly lipophilic drugs (LogP > 5) with high solubility in triglycerides (Cs > 50 mg/mL), ie, APIs that are candidates for lymphatic absorption.
NEW TRENDS IN LBDDS
Creation of Lipophilic Salts/Ion Pairs of Drugs for Solubilization in Lipidic Excipients
Despite the vast array of excipients to enable development of a LBDDS in which the drug is fully solubilized, some molecules will not dissolve in lipidic excipients at their required unit dose. While suspensions in LBDDS can provide good exposure and are commercially available, for example CiproTM Oral Suspension, typically, the best exposure from LBDDS is achieved when the drug is fully solubilized in the dosage form. In addition, formulation and manufacture of solutions present less challenges than suspensions that can be prone to aggregation and settling. The inability to solubilize an active agent in lipidic excipients has led formulators to rule out LBDDS as a viable technology for the drug. This unfortunate circumstance has restricted the use of this very versatile LBDDS approach to enable formulation of drugs with high formulation barriers, including poor bioavailability.
Recent work has been conducted to prepare lipophilic salts (or ion pairs) of drugs that enable increased and fully solubilized drug loads in lipidic excipients. Sahbaz and co-workers prepared docusate or decylsulfate salts of itraconazole, cinnarizine, and halofantrine to form ionic liquids or low melting point solids that were miscible or could be solubilized in SEDDS composed of long or medium chain triglycerides, surfactants, and cosolvents.72 Itraconazole docusate or cinnarizine decylsulfate were administered to rats in SEDDS formulations in which the dose was fully solubilized. The exposure of the fully solubilized drugs in the SEDDS formulation (made possible by the synthesis of lipophilic salts of these drugs) was 2-fold higher for cinnarizine and 20-fold higher for itraconazole, relative to control formulations of the suspended free base forms of the drugs at the same dose. This study demonstrated that formation of lipophilic salts or ion pairs could enable complete solubilization of drugs in lipidic excipients and greatly improve their exposure. This important study should start a paradigm shift in which less water-soluble and more lipid-soluble salt forms or ion pairs of drugs are synthesized to permit use of the LBDDS for drug delivery.
Use of LBDDS for Oral Delivery of Peptides & Proteins
Two recent papers focused on incorporation of large therapeutic peptides, namely leuprorelin, insulin, and desmopressin into SEDDS formulations consisting of CapryolTM 90, Labrafil® M 2125 CS, Labrasol® ALF, PeceolTM, propylene glycol, tetraglycol, Transcutol® HP, and Tween 20 at various compositions.72-73 Hydrophobic ion pairing of the peptides with anionic surfactants sodium docusate, dodecyl sulfate, and oleate was performed to enable solubilization of these highly water-soluble peptides into the oily SEDDS formulations. The hydrophobic ion pairs were evaluated by measuring the quantity of the complex formed, the n-octanol/water partition coefficient and the zeta potential of the complexes. For all three peptides, the ion pair formed with docusate gave the best combination of properties. It was observed that the docusate ion pairs of these peptides were soluble in all SEDDS formulations at greater than 10% by weight. Here, we see again, how pairing of compounds with lipophilic salts enables high loading of fully solubilized therapeutic agents into a LBDDS. As discussed in a previous section, the SEDDS protected the peptides against enzymatic degradation by intestinal proteases (α-chymotrypsin, trypsin, and elastase) and glutathione.72 Release of the peptides from the SEDDS formulations into 50-mm Tris buffer at pH 6.8 and 37°C was studied using a dialysis membrane method. All formulations permitted release of the peptides over 6 hours. These landmark papers suggest that LBDDS can be a viable formulation approach for oral delivery of peptides.
REFERENCES
- Thakkar H, Patel B, Thakkar S. A REVIEW ON TECHNIQUES FOR ORAL BIOAVAILABILITY ENHANCEMENT OF DRUGS. International Journal of Pharmaceutical Sciences Review and Research. 2010;4(3):203-223. doi:ISSN 0976 – 044X.
- Vasanthavada M, Serajuddin A. Lipid-Based Self-Emulsifying Solid Dispersions. In: Hauss D, ed. Oral Lipid-Based Formulations Enhancing The Bioavailability Of Poorly Water-Soluble Drugs. 1st ed. New York: Informa Healthcare; 2007:149-183.
- Carrier R, Miller L, Ahmed I. The utility of cyclodextrins for enhancing oral bioavailability. Journal of Controlled Release. 2007;123(2):78-99. doi:10.1016/j.jconrel.2007.07.018.
- Hauss D. Oral Lipid-Based Formulations. New York: Informa Healthcare; 2007:vii-viii.
- Williams H, Trevaskis N, Charman S et al. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacological Reviews. 2013;65(1):315-499. doi:10.1124/pr.112.005660.
- Jannin V, Chevrier S, Michenaud M et al. Development of self emulsifying lipid formulations of BCS class II drugs with low to medium lipophilicity. International Journal of Pharmaceutics. 2015;495(1):385-392. doi:10.1016/j.ijpharm.2015.09.009.
- Vemulapalli V, Khan N, Jasti B. Physicochemical Characteristics that Influence the Transport of Drugs Across Intestinal Barrier. AAPS news magazine. 2007;March:18-21.
- Lipinski C, Lombardo F, Dominy B, Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 1997;23(1-3):3-25. doi:10.1016/s0169-409x(96)00423-1.
- Barditch-Crovo P, Petty B, Gambertoglio J et al. The effect of increasing gastric pH upon the bioavailability of orally-administered foscarnet. Antiviral Research. 1998;38(3):209-212. doi:10.1016/s0166-3542(98)00024-2.
- Hetényi G, Griesser J, Moser M, Demarne F, Jannin V, Bernkop-Schnürch A. Comparison of the protective effect of self-emulsifying peptide drug delivery systems towards intestinal proteases and glutathione. International Journal of Pharmaceutics. 2017;523(1):357-365. doi:10.1016/j.ijpharm.2017.03.027.
- Haeberlin B, Gengenbacher T, Meinzer A, Fricker G. Cyclodextrins — Useful excipients for oral peptide administration? International Journal of Pharmaceutics. 1996;137(1):103-110. doi:10.1016/0378-5173(96)04499-7.
- O’Driscoll C. Lipid-based formulations for intestinal lymphatic delivery. European Journal of Pharmaceutical Sciences. 2002;15(5):405-415. doi:10.1016/s0928-0987(02)00051-9.
- Hauss D, Fogal S, Ficorilli J et al. Lipid-Based Delivery Systems for Improving the Bioavailability and Lymphatic Transport of a Poorly Water-Soluble LTB4 Inhibitor. Journal of Pharmaceutical Sciences. 1998;87(2):164-169. doi:10.1021/js970300n.
- Porter C, Charman W. In: Hauss D, ed. Oral Lipid-Based Formulations: Enhancing The Bioavailability Of Poorly Water-Soluble Drugs. 1st ed. New York: Informa Healthcare USA, Inc.; 2007:185-205.
- Charman W. Lipid vehicle and formulation effects on intestinal lymphatic drug transport. In: Charman W, Stella V, ed. Lymphatic Transport Of Drugs. 1st ed. Boca Raton: CRC Press; 1992:113-179.
- Porter C. Lymphatic transport of proteins after s.c. injection: implications of animal model selection. Advanced Drug Delivery Reviews. 2001;50(1-2):157-171. doi:10.1016/s0169-409x(01)00153-3.
- Porter C, Charman W. Intestinal lymphatic drug transport: an update. Advanced Drug Delivery Reviews. 2001;50(1-2):61-80. doi:10.1016/s0169-409x(01)00151-x.
- Uekama K. Design and Evaluation of Cyclodextrin-Based Drug Formulation. Chemical & Pharmaceutical Bulletin. 2004;52(8):900-915. doi:10.1248/cpb.52.900.
- Dahan A, Sabit H, Amidon G. Multiple Efflux Pumps Are Involved in the Transepithelial Transport of Colchicine: Combined Effect of P-Glycoprotein and Multidrug Resistance-Associated Protein 2 Leads to Decreased Intestinal Absorption Throughout the Entire Small Intestine. Drug Metabolism and Disposition. 2009;37(10):2028-2036. doi:10.1124/dmd.109.028282.
- Collnot E, Baldes C, Wempe M et al. Mechanism of Inhibition of P-Glycoprotein Mediated Efflux by Vitamin E TPGS: Influence on ATPase Activity and Membrane Fluidity. Molecular Pharmaceutics. 2007;4(3):465-474. doi:10.1021/mp060121r.
- Loo T, Clarke D. Recent Progress in Understanding the Mechanism of P-Glycoprotein-mediated Drug Efflux. Journal of Membrane Biology. 2005;206(3):173-185. doi:10.1007/s00232-005-0792-1.
- Williams H, Trevaskis N, Charman S et al. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacological Reviews. 2013;65(1):315-499. doi:10.1124/pr.112.005660.
- Brough C, Williams R. Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. International Journal of Pharmaceutics. 2013;453(1):157-166. doi:10.1016/j.ijpharm.2013.05.061.
- Liu H, Taylor L, Edgar K. The role of polymers in oral bioavailability enhancement; a review. Polymer. 2015;77:399-415. doi:10.1016/j.polymer.2015.09.026.
- Elder D, Holm R, Diego H. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. International Journal of Pharmaceutics. 2013;453(1):88-100. doi:10.1016/j.ijpharm.2012.11.028.
- Guerrieri P, Rumondor A, Li T, Taylor L. Analysis of Relationships Between Solid-State Properties, Counterion, and Developability of Pharmaceutical Salts. AAPS PharmSciTech. 2010;11(3):1212-1222. doi:10.1208/s12249-010-9499-4.
- Gould P. Salt selection for basic drugs. International Journal of Pharmaceutics. 1986;33(1-3):201-217. doi:10.1016/0378-5173(86)90055-4.
- Carrier R, Miller L, Ahmed I. The utility of cyclodextrins for enhancing oral bioavailability. Journal of Controlled Release. 2007;123(2):78-99. doi:10.1016/j.jconrel.2007.07.018.
- Connors K. The Stability of Cyclodextrin Complexes in Solution. Chemical Reviews. 1997;97(5):1325-1358. doi:10.1021/cr960371r.
- Yunomae K, Arima H, Hirayama F, Uekama K. Involvement of cholesterol in the inhibitory effect of dimethyl-β-cyclodextrin on P-glycoprotein and MRP2 function in Caco-2 cells. FEBS Letters. 2003;536(1-3):225-231. doi:10.1016/s0014-5793(03)00059-0.
- Uekama K. Design and Evaluation of Cyclodextrin-Based Drug Formulation. CHEMICAL & PHARMACEUTICAL BULLETIN. 2004;52(8):900-915. doi:10.1248/cpb.52.900.
- Baghel S, Cathcart H, O’Reilly N. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. Journal of Pharmaceutical Sciences. 2016;105(9):2527-2544. doi:10.1016/j.xphs.2015.10.008.
- Van den Mooter G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discovery Today: Technologies. 2012;9(2):e79-e85. doi:10.1016/j.ddtec.2011.10.002.
- Zhang M, Li H, Lang B et al. Formulation and delivery of improved amorphous fenofibrate solid dispersions prepared by thin film freezing. European Journal of Pharmaceutics and Biopharmaceutics. 2012;82(3):534-544. doi:10.1016/j.ejpb.2012.06.016.
- Kawakami K, Pikal M. Calorimetric Investigation of the Structural Relaxation of Amorphous Materials: Evaluating Validity of the Methodologies. Journal of Pharmaceutical Sciences. 2005;94(5):948-965. doi:10.1002/jps.20298.
- Dengale S, Hussen S, Krishna B, Musmade P, Gautham Shenoy G, Bhat K. Fabrication, solid state characterization and bioavailability assessment of stable binary amorphous phases of Ritonavir with Quercetin. European Journal of Pharmaceutics and Biopharmaceutics. 2015;89:329-338. doi:10.1016/j.ejpb.2014.12.025.
- Bhugra C, Pikal M. Role of Thermodynamic, Molecular, and Kinetic Factors in Crystallization from the Amorphous State. Journal of Pharmaceutical Sciences. 2008;97(4):1329-1349. doi:10.1002/jps.21138.
- Knapik J, Wojnarowska Z, Grzybowska K et al. Physical Stability of the Amorphous Anticholesterol Agent (Ezetimibe): The Role of Molecular Mobility. Molecular Pharmaceutics. 2014;11(11):4280-4290. doi:10.1021/mp500498e.
- Mistry P, Mohapatra S, Gopinath T, Vogt F, Suryanarayanan R. Role of the Strength of Drug–Polymer Interactions on the Molecular Mobility and Crystallization Inhibition in Ketoconazole Solid Dispersions. Molecular Pharmaceutics. 2015;12(9):3339-3350. doi:10.1021/acs.molpharmaceut.5b00333.
- Kothari K, Ragoonanan V, Suryanarayanan R. The Role of Polymer Concentration on the Molecular Mobility and Physical Stability of Nifedipine Solid Dispersions. Molecular Pharmaceutics. 2015;12(5):1477-1484. doi:10.1021/mp500800c.
- Broman E, Khoo C, Taylor L. A comparison of alternative polymer excipients and processing methods for making solid dispersions of a poorly water soluble drug. International Journal of Pharmaceutics. 2001;222(1):139-151. doi:10.1016/s0378-5173(01)00709-8.
- Ilevbare G, Liu H, Edgar K, Taylor L. Understanding Polymer Properties Important for Crystal Growth Inhibition—Impact of Chemically Diverse Polymers on Solution Crystal Growth of Ritonavir. Crystal Growth & Design. 2012;12(6):3133-3143. doi:10.1021/cg300325p.
- Vasanthavada M, Serajuddin A. Lipid-Based Self-Emulsifying Solid Dispersions. In: Hauss D, ed. Oral Lipid-Based Formulations Enhancing The Bioavailability Of Poorly Water-Soluble Drugs. 1st ed. New York: Informa Healthcare; 2007:149-183.
- Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discovery Today. 2011;16(7-8):354-360. doi:10.1016/j.drudis.2010.02.009.
- Yadav D, Kumar N. Nanonization of curcumin by antisolvent precipitation: Process development, characterization, freeze drying and stability performance. International Journal of Pharmaceutics. 2014;477(1-2):564-577. doi:10.1016/j.ijpharm.2014.10.070.
- Junyaprasert V, Morakul B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian Journal of Pharmaceutical Sciences. 2015;10(1):13-23. doi:10.1016/j.ajps.2014.08.005.
- Gauniya A, Mazumder R, Pathak K. Enhancement of Dissolution Profile of Poorly Water Soluble Drugs by Using Techniques of Nanocrystallization. Current Nanomedicine. 2016;6(3):240-250. doi:10.2174/2468187306666161021103832.
- Jia L. Nanoparticle Formulation Increases Oral Bioavailability of Poorly Soluble Drugs: Approaches, Experimental Evidences and Theory. Current Nanoscience. 2005;1(3):237-243. doi:10.2174/157341305774642939.
- Deshpande R, D. V. G, Vegesna N, Vaghela R, P. K. K. The effect of nanonization on poorly water soluble glibenclamide using a liquid anti-solvent precipitation technique: aqueous solubility, in vitro and in vivo study. RSC Adv. 2015;5(99):81728-81738. doi:10.1039/c5ra12678a.
- Savla R, Browne J, Plassat V, Wasan K, Wasan E. Review and analysis of FDA approved drugs using lipid-based formulations. Drug Development and Industrial Pharmacy. 2017:1-16. doi:10.1080/03639045.2017.1342654.
- Strickley R. Currently Marketed Oral Lipid-Based Dosage Forms: Drug Products and Excipients. In: Hauss D, ed. Oral Lipid-Based Formulations Enhancing The Bioavailability Of Poorly Water-Soluble Drugs. 1st ed. New York: Inform Healthcare USA, Inc.; 2007:1 -31.
- Fatouros D, Bergenstahl B, Mullertz A. Morphological observations on a lipid-based drug delivery system during in vitro digestion. European Journal of Pharmaceutical Sciences. 2007;31(2):85-94. doi:10.1016/j.ejps.2007.02.009.
- Pouton C. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. European Journal of Pharmaceutical Sciences. 2006;29(3-4):278-287. doi:10.1016/j.ejps.2006.04.016.
- Tuleu C, Newton M, Rose J et al. Comparative Bioavailability Study in Dogs of a Self‐Emulsifying Formulation of Progesterone Presented in a Pellet and Liquid form Compared with an Aqueous Suspension of Progesterone. Journal of Pharmaceutical Sciences. 2004;93(6):1495-1502. doi:10.1002/jps.20068.
- Khoo S, Humberstone A, Porter C, Edwards G, Charman W. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. International Journal of Pharmaceutics. 1998;167(1-2):155-164. doi:10.1016/s0378-5173(98)00054-4.
- Yin Y, Cui F, Mu C et al. Docetaxel microemulsion for enhanced oral bioavailability: Preparation and in vitro and in vivo evaluation. Journal of Controlled Release. 2009;140(2):86-94. doi:10.1016/j.jconrel.2009.08.015.
- Wei L, Sun P, Nie S, Pan W. Preparation and Evaluation of SEDDS and SMEDDS Containing Carvedilol. Drug Development and Industrial Pharmacy. 2005;31(8):785-794. doi:10.1080/03639040500216428.
- Jannin V, Chevrier S, Michenaud M et al. Development of self emulsifying lipid formulations of BCS class II drugs with low to medium lipophilicity. International Journal of Pharmaceutics. 2015;495(1):385-392. doi:10.1016/j.ijpharm.2015.09.009.
- Alskär L, Porter C, Bergström C. Tools for Early Prediction of Drug Loading in Lipid-Based Formulations. Molecular Pharmaceutics. 2016;13(1):251-261. doi:10.1021/acs.molpharmaceut.5b00704.
- Porter C, Trevaskis N, Charman W. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nature Reviews Drug Discovery. 2007;6(3):231-248. doi:10.1038/nrd2197.
- KOGA K, KUSAWAKE Y, ITO Y, SUGIOKA N, SHIBATA N, TAKADA K. Enhancing mechanism of Labrasol on intestinal membrane permeability of the hydrophilic drug gentamicin sulfate. European Journal of Pharmaceutics and Biopharmaceutics. 2006;64(1):82-91. doi:10.1016/j.ejpb.2006.03.011.
- Rama Prasad Y, Eaimtrakarn S, Ishida M et al. Evaluation of oral formulations of gentamicin containing labrasol in beagle dogs. International Journal of Pharmaceutics. 2003;268(1-2):13-21. doi:10.1016/j.ijpharm.2003.08.008.
- Chang R, Shojaei A. Effect of a lipoidic excipient on the absorption profile of compound UK 81252 in dogs after oral administration. J Pharm Pharm Sci. 2004;7(1):8-12.
- Rama Prasad Y, Minamimoto T, Yoshikawa Y et al. In situ intestinal absorption studies on low molecular weight heparin in rats using Labrasol as absorption enhancer. International Journal of Pharmaceutics. 2004;271(1-2):225-232. doi:10.1016/j.ijpharm.2003.11.013.
- Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. European Journal of Pharmaceutical Sciences. 2005;24(5):477-486. doi:10.1016/j.ejps.2005.01.001.
- Williams H, Sassene P, Kleberg K et al. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 1: Method Parameterization and Comparison of In Vitro Digestion Profiles Across a Range of Representative Formulations. Journal of Pharmaceutical Sciences. 2012;101(9):3360-3380. doi:10.1002/jps.23205.
- Williams H, Trevaskis N, Yeap Y, Anby M, Pouton C, Porter C. Lipid-Based Formulations and Drug Supersaturation: Harnessing the Unique Benefits of the Lipid Digestion/Absorption Pathway. Pharmaceutical Research. 2013;30(12):2976-2992. doi:10.1007/s11095-013-1126-0.
- Hetényi G, Griesser J, Moser M, Demarne F, Jannin V, Bernkop-Schnürch A. Comparison of the protective effect of self-emulsifying peptide drug delivery systems towards intestinal proteases and glutathione. International Journal of Pharmaceutics. 2017;523(1):357-365. doi:10.1016/j.ijpharm.2017.03.027.
- Hauss D, Fogal S, Ficorilli J et al. Lipid-Based Delivery Systems for Improving the Bioavailability and Lymphatic Transport of a Poorly Water-Soluble LTB4 Inhibitor. Journal of Pharmaceutical Sciences. 1998;87(2):164-169. doi:10.1021/js970300n.
- Charman W, Stella V. Transport of lipophilic molecules by the intestinal lymphatic system. Advanced Drug Delivery Reviews. 1991;7(1):1-14. doi:10.1016/0169-409x(91)90046-f.
- O’Driscoll C. Lipid-based formulations for intestinal lymphatic delivery. European Journal of Pharmaceutical Sciences. 2002;15(5):405-415. doi:10.1016/s0928-0987(02)00051-9.
- Sahbaz Y, Williams H, Nguyen T et al. Transformation of Poorly Water-Soluble Drugs into Lipophilic Ionic Liquids Enhances Oral Drug Exposure from Lipid Based Formulations. Molecular Pharmaceutics. 2015;12(6):1980-1991. doi:10.1021/mp500790t.
- Griesser J, Hetényi G, Moser M, Demarne F, Jannin V, Bernkop-Schnürch A. Hydrophobic ion pairing: Key to highly payloaded self-emulsifying peptide drug delivery systems. International Journal of Pharmaceutics. 2017;520(1-2):267-274. doi:10.1016/j.ijpharm.2017.02.019.

Dr. Jason LePree received his B.S. in Pharmacy from Rutgers University and his M.S. and Ph.D. in Pharmacy (Pharmaceutics) from the University of Wisconsin-Madison . He has over 20 years of experience in both Academia and Industry where he held leadership positions in preformulation, formulation, analytical R&D, and quality control. In parallel, Jason has served as adjunct professor at Long Island University since 1998, instructing on Physical Chemistry, Pharmacokinetics, and Pharmaceutical Analysis. Since joining Gattefossé in 2016, Jason has overseen the completion of the Gattefossé pharmaceutical application laboratories in Paramus, NJ. Equipped with the latest analytical, characterization, and processing capabilities, the laboratories’ mission is to help troubleshoot and advance customer projects to completion. Spearheading the support activity along with internal projects is the continuing mission of Dr. LePree.
Total Page Views: 26150



