


Issue:April 2018
ORALLY INHALED PRODUCTS - Optimizing the Application of In Vitro Test Methods for the Demonstration of Bioequivalence in Orally Inhaled Products
INTRODUCTION
Orally Inhaled Products (OIPs) are a commercially compelling target for generic development, with the combined annual revenues of key products, such as Seretide®, Spiriva®, and Symbicort® in the region of $10 billion.1 Diseases of the respiratory system account for 8% of all deaths in the EU, driving demand for treatments that are safe, efficacious, and cost effective. Replicating the performance of an OIP and demonstrating bioequivalence (BE) is complex, largely because OIP behavior is a function of interactions between the patient, device, and formulation. Ensuring the development of an optimal approach to the demonstration of BE is an important step in accelerating safe and effective generic products to market.
Demonstrating BE in any generic product typically relies on a combination of in vitro, pharmacokinetic (PK) and pharmacodynamics (PD) studies, though in vitro studies alone may be sufficient for certain products. For OIPs, there is ongoing debate as to the relevance of these different tests, and the Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidance differs in this regard. Reducing requirements for PK and/or PD studies is an attractive proposition because in vitro testing is typically the least expensive option. Maximizing the clinical relevance of in vitro test methods – an important and ongoing theme in OIP research – supports this goal.
The following examines the testing strategies demonstrating the BE of OIPs, their relevance, and the submission approaches outlined by the FDA and EMA. A key focus is the application of in vitro test methods and how these can be modified beyond the standard tests developed primarily for quality control (QC), to give improved in vitro in vivo correlations (IVIVCs) that are more useful for BE studies.
TESTING FOR BE
A generic product is prescribed interchangeably with the reference product and must therefore deliver closely equivalent clinical efficacy. In vitro tests, PK and/or PD studies are all routinely used to support claims of BE between a test (T) and reference (R) product, but differ markedly in terms of their: Complexity, Practicality, Discriminating Power, Clinical Relevance, and Cost. Assessing how inhaled drugs reach the lung and achieve therapeutic action helps to elucidate the relevance, value, and limitations of these different testing strategies, and highlights the unique difficulties associated with demonstrating BE between OIPs.
During inhalation, aerosolized particles are drawn from the OIP through the oropharyngeal region, into the main airways of the lung, and potentially into the deep lung. An upper size limit of 10 microns is typically assumed for penetration into the upper airways and 5 microns for deposition in the deep lung, with coarser particles depositing in the mouth/throat and likely entering the bloodstream via the gastrointestinal (GI) tract. When deposited in the deep lung, inhaled drug particles dissolve in the limited quantities of fluid that line the lungs, although mucociliary clearance (MCC) mechanisms simultaneously act to flush the particles from the body. Permeation into the lung tissue brings the dissolved drug into contact with its intended target, enabling localized binding and therapeutic action. Any drug absorbed through the lung tissue enters into systemic circulation (Figure 1).
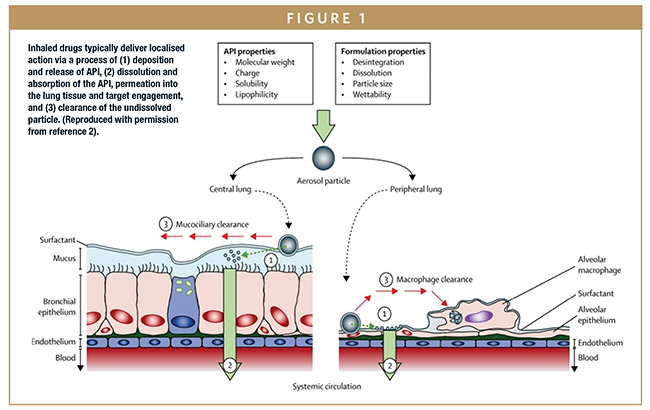
In vitro test methods are used to quantify a number of metrics directly associated with OIP efficacy. Core tests include the amount of drug delivered under standardized/well-controlled conditions – delivered dose uniformity (DDU) – and the aerodynamic particle size distribution (APSD) of that dose, which is measured using cascade impaction and influences in vivo deposition behavior. Other tests may include spray plume and plume geometry measurements, in the case of metered-dose inhalers (MDIs).
Easily repeatable and validated in vitro methods are relied upon for product QC. However, few good examples of IVIVCs exist for OIPs, due to factors such as variability in anatomy/impairment of the lung, device use, and compliance, which make it difficult to secure robust relationships between product characteristics and clinical efficacy. As a result, in vitro testing is often and necessarily supported by in vivo studies (PK/PD) for the demonstration of BE.
PK studies determine the fate of the drug substance within the body, primarily by tracking drug concentration in the blood plasma. PK studies are relatively straightforward to implement and can exhibit high discriminating power, particularly if a healthy patient population is used.3 It is generally accepted that if the systemic concentration measured for T is the same or less than that measured for R then systemic effects will be equivalent or less severe. This conclusion can mitigate the need for PD testing.3
The use of PK studies as an indicator of pulmonary bioavailability and hence, clinical efficacy, is more challenging because it can be argued that drug concentration in the blood is the result of pulmonary fate, rather than a reliable indicator of concentration/effect at the site of action in the lung.3-5 The implications for clinical efficacy of a difference in PK study results may therefore only be resolved via further in vivo studies.
PD studies quantify the biological and physiological impact of the drug substance and can be used to investigate both safety and efficacy with a high degree of clinical relevance. However, they can be relatively difficult to implement with a tendency to exhibit high variability and sensitivity. Successful implementation relies on the identification of a measurable, clinically relevant biomarker linked with the pharmacological mechanism of the drug. This biomarker must enable the demonstration of a dose-response relationship, when tested with representative doses, for a study to be differentiating. If T and R products result in a similar clinical endpoint, then this can only be indicative of similar efficacy if a change in dose is rigorously associated with a measurable response.4,5
THE REGULATORY LANDSCAPE FOR GENERIC OIPS
The complexities of PK/PD studies for OIPs, and the difficulty of establishing optimal strategies for their application in the demonstration of BE, are arguably reflected in the differences in approach in regulatory guidance in this area from the EMA and the FDA.
The stepwise approach for a generic OIP submission set out in the latest regulatory guidance released by the EMA indicates that a submission can be accepted on the basis of in vitro data alone (Figure 2) with no additional requirement for PK/PD testing.6
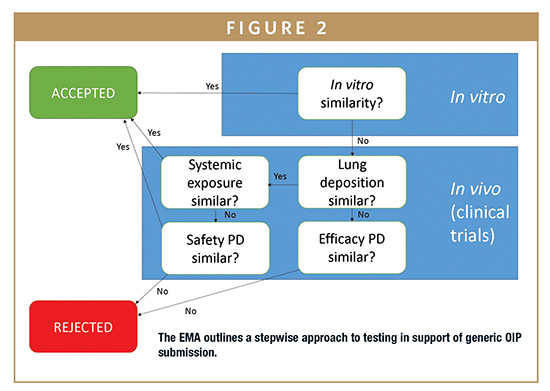
The elimination of PK/PD testing substantially streamlines the submission process and is highly appealing from the perspective of time and cost savings; however, few products have yet to be approved on the basis of in vitro test data alone. The criteria for demonstrating in vitro only BE are demanding and call for the test product to match not only the chemical and formulation characteristics of the reference product, but also for the devices and their behavioral characteristics to be highly similar. This can be severely limiting in practice, especially when commercial and intellectual property considerations are taken into account. Furthermore, the application of a battery of in vitro methods with the rigorous comparison of APSD measurements is a core element of such studies.6,7
The FDA has no comparable formalized guidance; however, the FDA 505(j) and 505(b)(2) pathways for generic and supergeneric OIP submissions, respectively, call for a quite different “weight of evidence” approach (Figure 3). This involves qualitative and quantitative formulation sameness, device similarity, PK (comparative systemic exposure studies) and PD studies, in addition to in vitro tests. Beyond this general guidance, the FDA also offers a steadily increasing number of product specific guidances for popular generic targets.8 These too typically indicate a requirement for both in vitro and in vivo (PK/PD) testing.
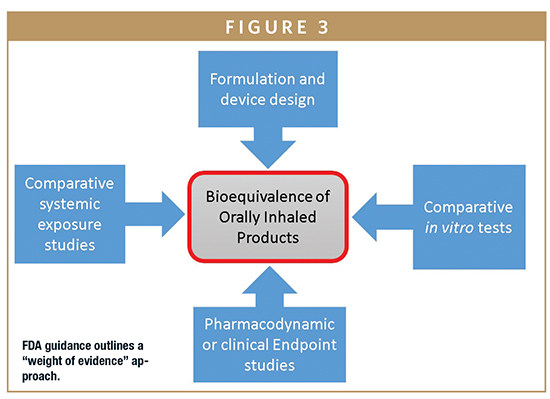
This current regulatory position is that most approved generic OIPs have been subjected to some form of in vivo testing. Such PK/PD studies not only add in time and cost but may also introduce additional risk, with poor IVIVC data complicating the demonstration of BE. Improving the clinical relevance of in vitro methods to access better IVIVCs and enable the greater reliance on in vitro methods for BE testing is an important goal for the industry.
MOVING TOWARDS BETTER IVIVCS
New models should be selected on the basis of their ability to reflect in vivo predictability – the primary aim – rather than anatomical correctness, where there is a choice to be made between the two.9 Ease of use and of production should also be considered. Simplifying models as far as possible without compromising predictability helps to realise this goal. Adopting these strategies has already resulted in the development of a number of products that can be used to improve IVIVCs, including:
-A more realistic throat model
-More representative breathing profiles
-The use of face models when testing add-on devices with face masks
-Dissolution testing
In a standard set-up for measuring the APSD of an OIP by cascade impaction (Figure 4), the product is interfaced to the impactor via the standard USP/Ph.Eur. induction port. This accessory has a simple right-angled geometry allowing reproducible drug recovery. However, it has been shown to capture less of the dose delivered by an OIP than would be deposited in the mouth-throat region during routine clinical use.10,11

From the perspective of anatomical representativeness, the use of throat cast is a solution to this issue. However, throat casts are patient specific, difficult to manufacture reproducibly, problematic to interface with the device/impactor, and have poor durability. The Alberta Idealised Throat (AIT) on the other hand is an alternative induction port (Figure 4) with a standard, idealized geometry developed from CT patient scans.
The AIT can be fully opened for drug recovery and to coat the internal surfaces to more closely simulate in vivo deposition. Adult and child versions are available for representative testing for specific patient groups. The AIT has been validated against clinical data over a period of around 10 years, and experimental data shows that it more accurately quantifies deposition than the standard USP/Ph.Eur. induction port.12 This accessory is therefore a good example of a practical design that delivers enhanced predictability.
More Representative Breathing Profiles
With many OIPs, the breathing maneuver of the patient directly influences drug delivery. These include dry powder inhalers (DPIs), where aerosol generation is typically driven solely by the inhalation maneuver of the patient, and nebulizers and MDIs with spacers/valved holding chambers (VHCs), which are operated with a tidal breathing pattern. Changes to the pharmacopoeial test methods for nebulizers, and more recently for MDIs with spacers/VHCs, reflect this with defined breathing profiles now specified to simulate product use by certain patient groups.13,14
Breath simulators are a cost-efficient solution for investigating how breath profiles impact drug delivery performance, with commercially available systems offering the flexibility to vary defining characteristics, such as inhalation or tidal volume, inhalation/exhalation ratio, frequency, and waveforms. Such studies help to elucidate the clinical efficacy that may be observed in different patient groups and are very much aligned with a Quality by De-sign (QbD) approach to product development and demonstration of BE. Indeed, confirming similarity between the performance of a T and R DPI across a range of flow rates supports claims that the products can be used interchangeably by all patients.
The Use of Face Models When Testing Add-On Devices With Face Masks
The correct use of MDIs requires the patient to begin to inhale immediately prior to actuation, thereby drawing the aerosolized dose directly into the lungs on the incoming breath. Certain patient groups, such as pediatrics, can find this level of coordination difficult to achieve and as a result, tend to use MDIs with spacers and VHCs. These inexpensive and easily retrofitted devices eliminate the need for coordination by providing a dead volume into which the dose is aerosolized, and from which the patient inhales the drug, by breathing tidally. However, the introduction of dead volume can impact both the amount and APSD of the delivered dose.
Newly released USP Chapter <1602> represents the latest thinking regarding the testing of MDIs with add-on devices and details test methods that are highly relevant for the robust demonstration of BE for MDIs.13 In cases where the spacer or VHC features a facemask, interfacing it with the test apparatus presents a significant challenge. The chapter allows for the use of face models that have the following clinical relevant characteristics:
-Appropriate facial dimensions for the intended user age range
-Ability to apply the facemask with the predicted amount of dead space when it is applied with a clinically relevant force to the model
-Physiological accurate soft facial tissue modelling around the chin, cheeks, and nose where the facemask makes contact
-Means of correctly mounting the spacer/VHC so that the facemask is oriented with the correct alignment to the face, as would occur when in use by the patient
Dissolution Testing
There are as yet no pharmacopoeial requirements for dissolution testing for OIPs, though this is clearly the subject of FDA interest.15 Where inhaled particles are very small then dissolution may be extremely rapid, however, for poorly soluble drugs, dissolution testing potentially has value for achieving a better understanding of in vivo behavior but remains challenging due to the limited amount and varying composition of fluid in the lung.
A number of methods have been proposed for dissolution testing, including the McConville/Copley methodology, which uses existing USP/Ph. Eur. tablet dissolution testing apparatus with sample captured using an NGI with modified cup and membrane holder (an alternative insert for the Andersen Cascade Impactor is also available).16 This set-up allows particles to be collected at defined impaction stages such that specific fractions of the APSD can be used for dissolution testing.
There is evidence that dissolution testing can distinguish between formulations of the same drug, and it is a particularly promising tool for investigating the performance of modified-release formulations, or poorly soluble drugs. However, there remain practical challenges to overcome in the development and application of suitable methodologies, not least in order to gather data that can be clearly correlated with in vivo behavior and clinical efficacy and that can consequently demonstrate of BE.
CONCLUSION
Demonstrating BE in OIPs requires a balance of simplicity, practicality, and reproducibility with clinical relevance. This is evident in issues associated with PK and PD studies and in moves to develop in vitro methods so as to improve their correlation with in vivo behavior. Improving clinical relevance often involves the introduction of complexity and increased variability. Greater variability translates into lower differentiating power, so a test that may be more clinically relevant for the demonstration of BE may be less able to detect a difference between a T and R product.
In vitro tests are the simplest of those to demonstrate BE, and their rigorous development toward better IVIVCs can help streamline generic OIP submissions. Optimizing the application of in vitro methods will help to cut the time and cost of generic development while at the same time ensuring the safety and efficacy of new products.
REFERENCES
1. Product Sales Data From Annual Reports Of Major Pharmaceutical Companies, Pharma Compass (2015). Website visited: (https://www.pharmacompass.com/pharma-data/product-salesdata-from-annual-reports-of-major-pharmaceutical-companies-2015).
2. Ruge CA, et al. Pulmonary drug delivery: from generating aerosols to overcoming biological barriers – therapeutic possibilities and technological challenges. The Lancet Respiratory Medicine. 2013;1(No. 5):402-413.
3. Evans C, et al. Equivalence considerations for orally inhaled products for local action – ISAM/IPAC-RS European Workshop Report. J Aerosol Med Pulm Drug Del. 2012;25(No. 3):117-139.
4. Adams WP, et al. Demonstrating bioequivalence of locally acting orally inhaled drug products (OIPs): Workshop Summary Report. J Aerosol Med Pulmo Drug Del. 2010;23(No. 1).
5. Al-Numani D, et al. Rethinking bioequivalence and equivalence requirements of orally inhaled drug products. Asian J Pharmaceut Sci. 2015;10(No. 6):461-471.
6. European Medicines Agency “Guideline on the requirements for clinical documentation for orally inhaled products (OIP) including the requirements for demonstration of therapeutic equivalence between two inhaled products for use in the treatment of asthma and chronic obstructive pulmonary disease (COPD) in adults and for use in the treatment of asthma in children and adolescents” Issued Jan 2009.
7. Forbes B, et al. In vitro testing for orally inhaled products: developments in science-based regulatory approaches. AAPS J. 2015;17(No. 4):837-852.
8. Product-Specific Guidances for Generic Drug Development. US Food and Drug Administration; Oct 2017. Website visited: (https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances/ucm075207.htm).
9. R Delvadia. Moving to more realistic in vitro testing of OIDPs. Presentation delivered at IPACRS/UF 2014 Conference. (http://ipacrs.org/assets/uploads/outputs/01_Day_2_OIC_2014_Delvadia.pdf).
10. Zhang Y, Gilbertson K, Finlay WH. In vivo – in vitro comparison of deposition in three mouththroat models with Qvar® and Turbuhaler® inhalers. J Aerosol Med Pulm Drug Del. 2007;20(No. 3):227-235.
11. Weers J, et al. In vitro–in vivo correlations observed with indacaterol-based formulations delivered with the Breezhaler. J Aerosol Med Pulm Drug Del. Website visited: https://www.ncbi.nlm.nih.gov/pubmed/25517988.
12. Copley M. Improving the realism and relevance of mouth-throat models for inhaled product testing. ONdrugDelivery Magazine. 2015;57:32-37.
13. Reviewer Guidance for Nebulizers, Metered Dose Inhalers, Spacers and Actuators, US Food and Drug Administration; 2015. Website visited: (https://www.fda.gov/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm081282.htm).
14. USP Chapter <1602> Spacers and Valved Holding Chambers Used with Inhalation Aerosols – Characterization Tests (2017).
15. Development of in vivo predictive dissolution method for orally inhaled drug products (U01), Department of Health and Human Services, National Institute of Health and US Food and Drug Administration;2013. Website visited: (http://grants.nih.gov/grants/guide/rfafiles/RFA-FD-13-014.html).
16. Copley M. Dissolution testing for inhaled drugs. Pharm Tech Eur. 2010;22(No. 11). Website visited: http://www.pharmtech.com/dissolution-testing-inhaled-drugs.

Mark Copley graduated from the University of Bath, UK, in 2000 with a Masters in Aerospace Engineering. For 8 years, he was Technical Sales Manager and Product Specialist for Copley Scientific’s range of inhaler testing equipment, before becoming the Sales Director in 2009. He is now Chief Executive Officer for the company. He is considered a leading authority in testing methods and systems for metered-dose inhalers, dry powder inhalers, nebulizers, and nasal sprays; authoring and contributing to more than 50 published articles. He also provides application support and consultancy, runs focused training workshops for the inhaled drug testing sector of the pharmaceutical industry, and sits on the editorial advisory panel of Inhalation Magazine. An invited member of the European Pharmaceutical Aerosol Group (EPAG) impactor subteam, he has also made recommendations to the Inhalanda working group, leading to subsequent revisions to Ph. Eur. And USP monographs. As part of Copley Scientific’s associate membership of the International Pharmaceutical Aerosol Consortium on Regulation & Science (IPAC-RS), he participates in a number of working groups with a view to enhancing the regulatory science of orally inhaled and nasal drug products (OINDPs).

Anna Sipitanou earned her BSc in Chemistry from the University of Bradford in 2014, and her MSc in Drug Discovery and Pharmaceutical Science from the University of Nottingham in 2015. She also undertook the role of Research Associate at the University of Nottingham, working to build in silico models for the prediction of drug induced liver injury. Building on previous experience at Cellomatics Biosciences, she joined Copley Scientific in July 2017 as Business Development Manager, playing a key role in the company’s service to customers, including training on a wide range of instruments.
Total Page Views: 9264



