


Issue:September 2018
CONTROLLED RELEASE - Informed Selection of Modified-Release Technologies Provides Simpler Oral Dose Regimens
INTRODUCTION
Controlled drug delivery (CDD) formulations are one of the favored tools for the lifecycle management of pharmaceutical products. Typically, a drug that is initially launched as multiple daily doses of an immediate-release (IR) product is reformulated into a once-daily modified-release product, simplifying dosing regimens while retaining efficacy and potentially improving its safety profile.
CDD formulations have many benefits, including extending the duration of effect, reducing toxicity that results from a high peak in plasma concentration, minimizing fluctuations in plasma concentration, and targeting drug release at a specific site within the gastrointestinal (GI) tract. Drugs with rapid clearance and short half-lives may require multiple daily doses and present adherence issues. Reducing frequency to once or twice daily dosing can lead to better patient adherence and acceptance.1
Delayed-release formulations are commonly used to release drugs in the correct part of the GI tract, notably drugs to treat colitis and Crohn’s disease, such as mesalazine.2 This approach can also protect an active pharmaceutical ingredient (API) that is sensitive to digestive acids or enzymes, or those that can cause gastric irritation.
Several drugs have a combination of immediate release with a controlled-release profile. This ensures rapid onset and sustained effect. The extended release (ER) formulation of the sedative-hypnotic, zolpidem, gives a biphasic absorption profile, with an IR component, inducing rapid onset of sleep; and then an ER maintenance dose gives a longer duration of action. As the API’s elimination half-life is less than 3 hours, it does not accumulate to cause daytime sleepiness.3 Similarly, the ER formulation of the attention-deficit/hyperactivity disorder (ADHD) drug, methylphenidate, has an IR coating over an osmotically active CR core to give rapid onset plus a 12-hour duration of action. Before investing time and resources to develop a CDD formulation, thorough drug characterization should be performed, and the data should be used for feasibility assessment. There are many important factors to determine if a CDD system is feasible and to help guide development. The following reviews some of the physicochemical and biopharmaceutical characteristics of drugs and how they influence creation of a CDD formulation.
ASSESSING FEASIBILITY & RISKS
Careful preparation and thorough understanding of drug properties underpin successful formulation and dosage form development and minimizes costs, risks, and time. Many factors contribute to the success or failure of CDD systems, as listed in Table 1.4,5 To achieve success, the fundamental physical and chemical characteristics of the API should be studied and applied, both on its own and in combination with proposed excipients to assess the feasibility of a CDD system. Physicochemical characterization should focus on the expected dosage forms and be designed to predict processing problems. For speed, minimal quantities of API should be consumed, and readily available analytical instruments are preferable.6
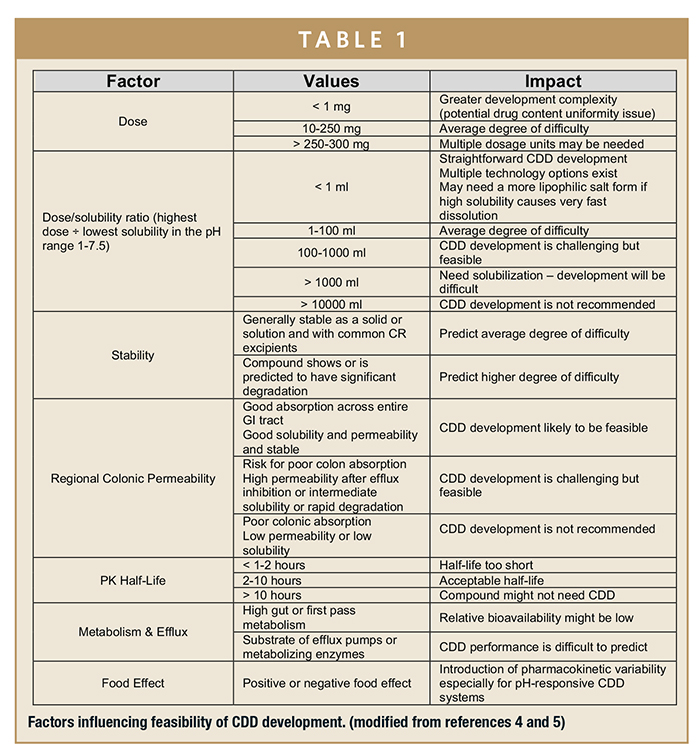
DOSE
Ideally, the dose will be in the range of 10 to 250 mg, which is large enough for uniformity not to be an issue, yet small enough to make a single, swallowable dosage form. If large amounts of excipients are necessary, this may require manufacturing of large tablets and capsules and present swallowability challenges.7 To avoid large dosage units, patients may need to take multiple tablets or capsules.
SOLUBILITY & DISSOLUTION RATE
Perhaps the most important parameter for CDD development is adequate solubility across the physiological pH range, particularly in relation to the dose. The drug should remain solubilized throughout the GI tract and not precipitate out. Table 1 shows that if the entire dose is soluble in 1 to 100 ml of GI fluid, the drug should present an average level of difficulty to formulate as a CDD. In contrast to IR formulations, which are predominantly absorbed in the upper GI tract, CDD products are more likely to also be absorbed in the distal small intestine and colon, and time to reach maximum plasma concentration may not be reached for at least 18 hours after dosing. If once-daily dosing is to be achieved, the drug will need to be soluble in the higher pH of the colon and also absorbed in the colon (see section on permeability).
Whatever the format of the CDD system (matrix, osmotic pump, layered tablet, coated systems), the system must also retain an appropriate dissolution rate throughout the physiological pH range. If a drug dissolves very rapidly, a CDD format is unlikely to be successful; one that dissolves very slowly may not need CDD. Historical applications of CDD focused on decreasing the dissolution rate of very soluble APIs to achieve constant plasma levels, perhaps by using a salt form with lower solubility. Granulation with lipophilic excipients, such as stearic acid, high-molecular weight polyethylene glycol (PEG), or white wax, have been used to slow down the dissolution rate. Drug release from these matrices is partially dependent on the excipient digestion. Because of differences in digestion processes between individuals, this approach can introduce inter-patient variability. New molecular entities (NMEs) in the current pharmaceutical pipeline tend to be more lipophilic and lower solubility. For these molecules, solubility enhancement must be combined with CDD or targeted delivery to attain adequate bioavailability and pharmacokinetics (PK) for CDD applications.
POLYMORPHISM & SALT FORMS
Solid state properties, such as polymorphism and salt formation, influence solubility and dissolution rate. Polymorph screening is an important step in a preformulation campaign, as it is estimated half of all organic molecules with a molecular weight below 600 have true polymorphs, and if solvates are counted, then it may be as high as 87%.8
Solubility can be tailored by changing the salt form if the API is ionizable. For a CDD product, the aim is to control solubility and dissolution to ensure prolonged release. If a drug is highly soluble, a salt form that reduces solubility may be advisable. Conversely, a salt may be used to improve solubility for those where it is very poor. Solubility enhancement will almost certainly still be required as part of the CDD system, but possibly not as much as would be necessary for an IR product.
PARTICLE SIZE
Drug particle size can affect parameters, including dissolution rate, and provide challenges for content uniformity, stability, and processing. Micronization can reduce particle size and improve uniformity, but micronized particles tend to agglomerate, particularly if they have a high surface energy. This can be minimized by specifying minimum particle size or adding excipients, such as silicon dioxide. Co-micronization is a technique in which pure drug is blended with a small amount of surfactant and micronized. This approach may improve particle wettability, very modestly improve solubility, and overcome processability issues seen in micronization. Homogeneity of a final product is more difficult to achieve when mixing ingredients with very different particle sizes, so size distribution may need to be carefully controlled.
PERMEABILITY
Once dissolved in GI fluid, drugs must have high permeability to be absorbed in the bloodstream by paracellular or transcellular routes, or both. The former is the main route for hydrophilic molecules, while the latter can be passive or carrier-mediated. While CDD development is of average difficulty for passive routes, it is harder to predict where other mechanisms, such as metabolism in the gut, are in play, or if it is subject to efflux.
A CDD must remain permeable throughout the GI tract, including the colon, and colonic permeability must be part of a feasibility assessment. Caco-2 cell permeability studies are routinely carried out, and there is some correlation to human colonic permeability.9 The higher levels of anaerobic bacteria in the colon makes it more difficult to correlate in vitro studies of colonic stability with in vivo behavior.
STABILITY & EXCIPIENT COMPATIBILITY
The stability of a drug decreases when combined with excipients, and continues to decline as drug concentration falls. There is no standard method for testing drug-excipient compatibility, and most methods are poor at predicting the final dosage form’s stability. Poor sample uniformity, unrealistic ratios of drug and excipient, the exclusion of key excipients, and the compatibility of testing methods with mixtures of excipients, can all limit their accuracy.
The ICH Q1A guideline lays out the required stability data package for a new drug.10 Its aim is to provide evidence of how environmental factors, such as temperature, humidity and light affect drug quality, shelf-life, and recommended storage conditions.
Studying stability both in solution and the solid state, and compatibility with excipients, is an important part of the Quality by Design (QbD) process, and forced degradation studies in both solution and solid state are very useful when determining optimal manufacturing and processing conditions. For example, if it degrades at high temperature, hot melt extrusion is not appropriate. Chromatography techniques are the most sensitive for determining stability; PXRD, infra-red and Raman spectroscopy, and nuclear magnetic resonance less so.
When selecting excipients for testing, the first step should be a literature search to identify any known incompatibilities between the drug’s functional groups and the excipient. Then, the compatibility of a single excipient is assessed at ratios that simulate finished dosage form. These studies take time – typically the combinations are stored in open bottles at 40°C and 75% relative humidity for 14 to 28 days, with the drug alone as a control, and then high-performance liquid chromatography (HPLC) used to quantify potency and impurities. Any excipients showing any incompatibility can be eliminated. Water may also be added to samples as moisture can trigger degradation.
MODELING DRUG METABOLISM & PHARMACOKINETICS
CDD formulations essentially work by altering drug release from the formulation and therefore the pharmacokinetics. As CDD formulations will have extended-release properties, the drug will be absorbed more slowly, the time to maximum concentration will be longer, and peak plasma concentration will be lower. Yet the area under the curve (AUC) should be the same for both IR and CDD products. Time to reach maximum plasma concentration is important for a CDD dosage form, as if it is reached quickly, this will not provide a successful once- or twice-daily dosing regimen. Similarly, if maximum plasma concentration is high, it may indicate that the formulation is inadequate because the drug is being released too rapidly.
The nature of CDD products increases the likelihood of food interactions, making APIs with significant food effects more difficult to formulate. The food effect introduces PK unpredictability, and makes it more difficult to achieve the desired plasma profile. Also, a CDD product that relies on pH for drug release may not perform as planned when co-administered with food, as this increases stomach pH, followed by increased gastric secretion and gastric emptying, which lowers duodenal pH. A “take on an empty stomach” label may be required.
PK modelling tools can be applied to assess the likelihood of CDD being viable by conferring PK/PD advantages. A range of commercial modelling and simulation tools are available to predict plasma profiles based on in vitro dissolution data, dose, and clearance. They can also be used to deconvolute data from PK studies.
The power of PK modelling is greatly influenced by both the amount and quality of data available. Early in the development process for a NME, much of the useful data may not be available, or only exist in preclinical species. Estimates may need to be made, based on similar molecules. When a CDD product is a follow-on from an IR dosage form, these data are likely to be known already. Data important to create accurate PK models largely reflect those discussed earlier and include dose, solubility, permeability, half-life, site of absorption, therapeutic window, and pH sensitivity. Data on food effect, logP, pKa, solubility in fasting and fed state simulated intestinal fluids (FaSSIF and FeSSIF), unbound fraction in human plasma, ratio of blood to plasma, intrinsic clearance, and fraction absorbed are also useful.
CONCLUSION
Ultimately, the successful development of a CDD product relies on a comprehensive preformulation strategy to characterize the molecules. The information gleaned in these studies will inform feasibility assessments, and minimize the risks, costs, and time in developing the product, greatly improving the chances of success.
REFERENCES
- Coleman CI, Limone B, Sobieraj DM, Lee S, Roberts MS, Kaur R, et al. Dosing frequency and medication adherence in chronic disease. J Manag Care Pharm. 2012;18(7):527-39.
- Lautenschlager C, Schmidt C, Fischer D, Stallmach A. Drug delivery strategies in the therapy of inflammatory bowel disease. Adv Drug Deliv Rev. 2014;71:58-76.
- Kirkwood C, Neill J, Breden E. Zolpidem modified-release in insomnia. Neuropsychiatr Dis Treat. 2007;3(5):521-6.
- Thombre AG. Assessment of the feasibility of oral controlled release in an exploratory development setting. Drug Discov Today. 2005;10(17):1159-66.
- Zhang HS, J.M. Biopharmaceutic Consideration and Assessment for Oral Controlled Release Formulations. In: Wen HP, K., editor. Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice. Hoboken, NJ: John Wiley & Sons, Inc.; 2010. p.33-45.
- Chrzanowski F. Preformulation considerations for controlled release dosage forms. Part I. Selecting candidates. AAPS PharmSciTech. 2008;9(2):635-8.
- Fields J, Go JT, Schulze KS. Pill Properties that Cause Dysphagia and Treatment Failure. Curr Ther Res Clin Exp. 2015;77:79-82.
- Hilfiker R. Polymorphism: in the pharmaceutical industry: John Wiley & Sons; 2006.
- Yee S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man–fact or myth. Pharm Res. 1997;14(6):763-6.
- https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2__Guideline.pdf.

Dr. Ronak Savla is a Scientific Affairs Manager with Catalent Pharma Solutions and the Catalent Applied Drug Delivery Institute. Previously, he was the Applied Drug Delivery Fellow at Catalent. He earned his PharmD and PhD from Rutgers University, New Jersey. His current research interests are the application of in silico models and simulations to aid in drug formulation design, integration of novel formulation technologies into the industry, and patient-centric research. Dr. Savla is an author and coauthor of more than 35 publications.
Total Page Views: 8786




