



Issue:January/February 2015
SPECIAL FEATURE - Analytical Testing: A Critical Element in Drug Development
There is intense competition within the pharmaceutical market – both with regards to developing cutting-edge therapies and in getting them through development quickly to take advantage of patent exclusivity. One of the critical elements in the drug development process is analytical testing. “Analytical testing is required at all phases of development, and it is critical that this support is accurate and efficient,” says Eric J. Hill, MS, MBA, President, Impact Analytical. “It is used to verify drug chemistry, and therefore can be a barrier to moving to the next phase in the development process.”
The importance of analytical testing is evident by the growth of the CRO sector. ISR’s report, “2014 CRO Market Size: 2012-2018,” points out that the CRO market was projected to reach $23.6 billion in 2014. With a 7.9% CAGR from 2014 to 2018, CROs are benefiting from the increased rate of outsourcing and increasing R&D budgets from biotech and specialty/generics companies.
This annual report from Drug Development & Delivery magazine highlights some of the leading players in the analytical testing space and how they are working with pharma clients to develop quality formulations, get drugs to market faster, and reduce risk.
ABC LABORATORIES—ADDRESSING CRITICAL ROLE OF CMC IN BIOPHARMACEUTICAL IND
Early-stage drug developers generally have their sights firmly set on the initiation of clinical trials, and rightly so: the outcomes of toxicological and pharmacological studies generally determine whether a drug candidate is viable for development. But as many pharmaceutical innovators have learned, the importance of Chemistry, Manufacturing and Control (CMC) data should not be underestimated. Insufficient data or a poorly thought-out approach to CMC can be pivotal to IND approval.

Meeting FDA expectations for CMC requires careful planning. This is particularly true of biopharmaceuticals due to their inherently complicated structures, multiple degradation sites, and cell culture impurities. While the FDA does not require complete characterization of the biopharmaceutical at the IND stage, it does expect a wide variety of issues to be addressed in a submission, such as identification of protein sequence, an activity assay based on binding, assays to demonstrate safety, purity and stability of drug substance and drug product, and determination of immunogenicity and the presence of anti-drug antibodies (ADA) in plasma, a safety concern unique to biopharmaceuticals that presents an additional analytical challenge.
“The diversity of CMC requirements for a biopharmaceutical IND present a variety of analytical challenges, and they require a myriad of analytical methods,” says Glenn Petrie, Ph.D., Senior Scientific Advisor, ABC Laboratories. “Many drug developers are overwhelmed by the multitude of options and decisions that must be made. Consequently, they often tend towards ‘analysis by paralysis.’ Rather than prioritize, they perform each and every study reasoning that they have covered all the bases relative to FDA requirements. This results in a tremendous waste of time and resources. Perhaps more importantly, the inclusion of superfluous data in a submission can lead to confusion and delays in the approval process.”
How can a drug developer ensure a safe, pure, and stable drug in the most efficient manner possible? Dr. Petrie suggests that studies need to be phase-appropriate, performed in parallel and proceed as a continuum. Up to and including Phase 1, analytical methods need only be qualified, not validated. Linearity, precision, accuracy, and specificity will suffice at this stage. When planning an approach to analytical methods, it is important to evaluate several possible techniques simultaneously and determine which are optimal for your application. Finally, it is important to approach analytical development as an iterative process of continuous refinement so that the improved/validated techniques are completed as the drug progresses through Phase 2, Phase 3, and commercialization.
AVOMEEN ANALYTICAL SERVICES—STAFF & EQUIPMENT DEVELOP RELIABLE TESTING METHODS
Modern CROs have to evolve to meet the growing needs within the pharmaceutical industry. As products become more complex the required analytical testing associated with them also builds in complexity, thus newer more sophisticated methods and instrumentation are required to meet ever increasing demands. The skills of a company that can properly meet these needs are strongly desired by many companies within the industry and are needed more and more every day.
“It’s interesting to see that problems that were once considered impossible not too long ago are now becoming realities in the pharma world, although the burden is on analytical chemists to provide safety easements and to develop reliable release testing methods for these products,” says Shri Thanedar, Ph.D., CEO & Chief Chemist of Avomeen Analytical Services. “One example of this is targeted drug delivery to a specific part of the body. This modern technique requires the development of a formulation that can not only successfully navigate and travel to a specific part of the body, but can do so without destroying or harming the API it contains.”
Dr. Thanedar explains that Avomeen assisted a client by developing an analytical method that tests for an otherwise insoluble component that the FDA required to be analyzed due to a particular ingredient. “We have developed an extraction and functionalization that is capable of quantifying that specific component,” he says. “This ability to solve for unknowns, overcome complex problems, and the utilizing of a unique blend of experienced staff from multiple industries and disciplines was critical to the success of this project.”

“What was once innovative just a few years ago is now considered routine and the level of sophistication needed to solve these modern problems grows daily,” continues Dr. Thanedar. “Much of the low-lying fruit is gone and companies have to reach farther to get a product on the market.”
To address its clients’ complex needs, Avomeen relies on its experienced staff and has a completely redesigned, customized 2,500-sq. ft. facility equipped with many segregated laboratories that contain state-of-the-art equipment, including GLP/GMP labs, R&D labs, formulation suites, ISO-7 compliant clean rooms, and more.
CATALENT PHARMA SOLUTIONS—OPTIMIZING SOLID-STATE FORMS
Programs in the early stages of development are often challenged by complexities associated with the chemical and physical properties of the drug. These properties can, in turn, lead to challenges developing analytical methodologies and delivering the drug in a manner that enables its safety and efficacy to be clearly established. Thus, it is recommended to partner with workforces comprised of highly trained scientists and analytical testing and development partners who have accumulated extensive libraries of experiences that span a range of molecule diversity and attributes. Many of these organizations have partnered with hundreds of companies to advance molecules over both technical and regulatory hurdles. Cultivated from these experiences are sophisticated and efficient workflows that drive fundamentally sound technical outcomes.
One example of this is Catalent Pharma Solutions’ Optiform® technologies platform. This platform combines sophisticated informatics and automation tools to design and assemble salt and crystal-form screening studies. “When combined with high-end solid-state analytical capabilities (e.g., high-throughput FTRaman, X-ray powder diffraction, and thermal analysis) and a highly experienced technical workforce, an optimized solid-state form can be selected in a short period of time utilizing a relatively small quantity of drug substance,” says David Igo, Director Product Development with Catalent. These materials can then be employed in early PK studies to support a fast-to-fail development model.

Development organizations draw their strength from an integration of services that begin with preformulation activities and extend through clinical and commercial manufacturing support. Through this integration, they are able to develop both broad and deep partnerships with clients that are supported by comprehensive analytical services. The services critical to the success of development programs include an agnostic preformulation service that allows the physico-chemical properties of the active pharmaceutical ingredient to be evaluated against attributes important to the intended product profile (e.g., solid-oral dosage form, subcutaneous injection) without being biased by a target drug-delivery technology.
“At Catalent, our technical experts have developed a screening tool (FormProRxTM) that considers the physical properties of the molecule along with its solubility and permeability attributes and handling requirements when proposing solid oral drug delivery,” says Dr. Igo.
IMPACT ANALYTICAL—A THREE-PRONGED APPROACH TO QUALITY RESULTS
Intense competition among drug developers has placed pressure on analytical testing providers to generate quality results quickly. Impact Analytical has responded to this pressure by assembling an experienced staff, equipped them with top-notch equipment, and designed a laboratory around its work process. The team of scientists is experts in their respective areas, such as the latest cGMP regulations and USP methods. “Having this experienced team ensures efficient transfer of knowledge between the development scientists and the analytical team,” explains Eric J. Hill, MS, MBA, President, Impact Analytical. “It also ensures the right experiments are undertaken to meet the demands of development, while meeting all regulatory requirements. If problems or unexpected results arise, an experienced team can react quickly without compromising the overall timeline.”
Just as important as staff is access to up-to-date equipment that is properly qualified and maintained. The Impact Analytical lab contains multiple UPLC, HPLC, GC, GC-MS, and LC-MS systems. “This gives us the bandwidth to handle large project loads, while ensuring the equipment can handle the demands of regulatory work,” says Mr. Hill. “We attempt to dedicate systems to common set ups (eluent sets, carrier gases, etc.), which decreases set-up time.”

Finally, lab set-up can be a contributing factor to throughput. Mr. Hill explains that Impact Analytical has deviated from the traditional setup of segregating analytical disciplines in separate laboratories (LC, GC, molecular characterization, etc.). “When we built our new facility, we created two main laboratories–a prep laboratory and an instrument laboratory. This reduces contamination by segregating chemicals and standards away from instruments. It also creates a better team atmosphere that promotes instrument sharing and cross-training. This set up matches our work processes, which improves throughput, safety, and accuracy.”
METRICS CONTRACT SERVICES—STAYING AHEAD OF NEW USP TESTING GUIDELINES
The pharmaceutical industry is facing substantive changes being made by the U.S. Pharmacopeial Convention (USP) concerning the testing of elemental impurities. The new guidelines take effect in December 2015.
Until recently, testing for metal impurities in pharmaceutical products has been limited to a few compendial procedures in excipients, residual catalysts, and a metal detector at the end of the production line to check for shards. That will change with the implementation of the USP’s new guidelines. The intent is to replace the existing benchtop, heavy-metals procedure with more modern and effective techniques, such as ICP and ICP-MS. The guidelines also introduce the requirement to monitor finished product for possible metal shed from processing equipment and container closures. Once the rollout is complete, manufacturers will be expected to have results for their products, as well as some assessment of process to assure minimal contamination.
Theoretically, testing for elemental impurities is like any other analytical procedure – the sample is prepared, tested on an instrument versus calibration standards, and the levels of impurities are calculated and reported. But practically, it requires specialized equipment for both preparation and analysis that many pharmaceutical labs do not currently operate.
Microwave digestion is the sample preparation tool of choice for ICP and ICP-MS. It is fast, effective, uses minimal sample, and is applicable to nearly all kinds of samples. Metrics offers different models that can be selected based on the required throughput. Sequential or batched digestion is available.
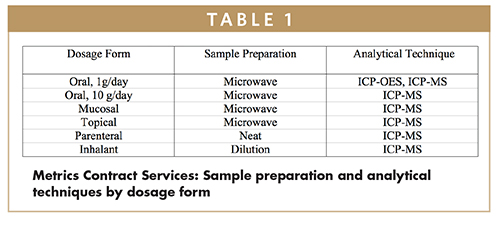
Once digested, the analyst will select ICP-OES or ICP-MS based on the sensitivity required. Because the required limits are scaled by the daily dose, ICP-MS will be used for the majority of analyses, especially for injectable or high-dosage products. The latest ICP-MS systems also use interference correction systems to reduce the risk of false positives. Conveniently for the analyst, both ICPOES and ICP-MS can determine more than 50 elements simultaneously.
Metrics operates a dedicated trace elements laboratory. “We currently operate two microwave digestion systems as well as late-generation ICP and ICP-MS instruments, and can leverage existing methodology to diverse products and dosage forms,” says Krzysztof “Chris” Golebiowski, Vice President of Analytical Services at Metrics Contract Services. “While these new requirements make a step into unfamiliar territory for a number of manufacturers, Metrics is positioned well ahead of the implementation of the new chapters with the experience and equipment that is needed.”
PARTICLE SCIENCES—SHORTENING TIMELINES & MINIMIZING RISKS
The Analytical Services Department at Particle Sciences consists of 21 dedicated scientists trained to characterize drug products at stages of drug development from proof-of-concept through support of clinical trial material release and stability testing. The group works in GMP-compliant laboratories in cross-functional teams led by internal project management. Combined, the group has more than a century of pharmaceutical analysis experience with small-molecule APIs and complex delivery systems for biologics. Using state-of-the-art instrumentation including HPLC, particle size analysis, mass spectrometry, rheology, gel electrophoresis, and other modern analytical techniques, the group provides timely and critical information on the drug product to guide formulation development, explains Laurie Goldman, Director, Analytical Services for Particle Sciences.
Assessment includes physical testing as well as chemical characterization on a range of dosage forms, including solid oral dosage, topical/mucosal/transdermal delivery, nanoparticulate and microparticulate injectables, ocular administration, and combination drugeluting devices. Methods are developed with the intent of providing immediate feedback on stability and delivery of drug. As such, methods are developed to be stability indicating and indicative of in-vivo release.
PSI uses stability-indicating methods in early stages of development to correlate how the manufacturing process influences the stability of the API. These results inform the formulators in their choices and direction. In later stages of development, stability-indicating methods are used in ICH-compliant long-term product stability studies. In vitro release studies are used in early stages of development to compare delivery from various dosage forms and formulations. In later stages of development, they may be used as quality control tests to indicate if changes in the product over time have resulted in any change to the release of drug substance from the drug product. Once in-vivo data is available, an in-vitro method that has been established to show in vivo/in vitro correlation can be used to assess potential changes to the drug product or manufacturing process.
Methods are developed to be fit-for-purpose, providing a level of confidence in the results produced from them. When appropriate, methods are validated according to ICH Q2B guidelines to a level appropriate to the development phase being supported. Physical characterization methods are performance-verified for support of GMP studies. In early stages of product development, when validation is not necessary, a more limited evaluation of analytical methods is performed to assure results generated from them are scientifically sound.
Compliance to GMP regulations is mandatory in all laboratories. Compliance is ensured by a combination of ongoing training of analysts on the techniques in use, formal qualification of instrumentation, and method validation. “These fundamentals provide the foundation on which solid, reliable methods are used to generate and interpret the critical information necessary for efficient and effective drug product development,” she says.
SGS LIFE SERVICES—A FOCUS ON BIOLOGICS
About 10 years ago, SGS Life Science Services took a step towards large molecules by opening its first biologics testing laboratory in Wavre, Belgium, and continues to invest in biologics testing. This was achieved through both the acquisition of the MScan Group and through the development of specific, large molecule testing services in a number of the existing, traditionally small molecule, SGS laboratories in its global network.
“For companies new to the biopharmaceutical market, they see SGS very much as a consultative partner rather than a service provider, and look to us for advice as to how to design experiments and how to deal with regulatory issues, whereas previously clients would approach us with a pre-determined list of tests to conduct,” says Dr. Mark Rogers, Senior Vice President, Life Science Services, SGS North America Inc. To reduce development times, especially amongst smaller and mid-size biotech companies, it is becoming more common for clients to engage with service providers earlier and on a more collaborative basis.
“The industry has the same types of challenges it always had, but technology has moved on and continues to influence the rate of change,” explains Dr. Rogers. “Clients, although all working in the same “industry,” will always have unique requests based on their experience and particular field of research. There is always the potential for a client to have an unexpected and immediate need for a unique test or expertise. It is up to service providers to anticipate the industry’s needs.”
Such an example is in the specific area of biologics testing. Dr. Rogers says that clients look for service providers to define higher order structures as the rule, rather than the exception. Such techniques are now applied in characterization of primary products, as well as for comparability studies such as in the testing of biosimilars. “This service has been offered by SGS for several years but has recently seen increased numbers of requests,” says Dr. Rogers. “Consequently, SGS has made investments in its capacity within its global laboratory network. The challenge will continue to be meeting the demand for increased rate of testing, with greater orders of precision and accuracy.”
SGS recently opened a facility in Carson, CA, to provide bioanalytical testing, its first facility in North America. Sites in France and Belgium have validated more than 700 methods and this experience can be transferred to Carson for clients who would be looking to have this testing performed in North America.
XCELIENCE—DEVELOPING ANALYTICAL DATA FOR BETTER FORMULATIONS
Quality Control is an important part of release testing for both commercial and clinical drug products. The accurate quantitation of drug in the dosage form is critical to ensure that the patient is neither under or over dosed. In addition, during formulation development, it is necessary to provide accurate testing results to help guide the formulators in the development and optimization of the dosage form. Better formulations and processes are proposed and defined based on the analytical data.
“Xcelience formulators work closely with the analysts to generate data that can be used to optimize the formulation,” says Yide Chang, Ph.D., Manager, Analytical Services, Xcelience. “The goal is to provide a simple, accurate and robust testing method that provides the necessary information. The analyst must always balance maintaining quality with the need for speed and cost.”
The principle types of analytical methods for new drug development are potency of the drug, impurities, and dissolution in various media. For potency and impurities, scientists develop simple and robust methods for their intended uses, which include monitoring stability of new drugs or new formulations. Method parameters and appropriate sample preparation procedures are critical in the development of new methods. For dissolution testing, the analytical team will develop suitable methods for water soluble or for highly soluble active pharmaceutical ingredients. Initially, the early methods will be used to compare different formulations. These methods should be developed well enough to provide formulators direction for decision making. Suitable analytical methods need to be well developed and evaluated.
Once the formulation is finalized, it will be a simple process to validate the analytical methods. Later, the methods will be used for quality control, performance of drug products in stability studies, IVIVC, or other purposes. “Xcelience’s expertise and focus during development has been extremely advantageous in our later activities for manufacturing for clinical and for commercial production,” says Paul Skultety, PhD, Vice President, Pharmaceutical Development Services, Xcelience. “Robust development results in test methods that can deliver accurate results with less chance for repeated testing, out of specification or out of tolerance results or other testing failures, as well as analytical results that can be reported with a high level of confidence.”
One of the biggest challenges for the Xcelience analysts is that there are more potent compounds in early development that require dosage strengths as low as microgram levels. These formulations present a challenge to the analyst to accurately quantitate the active compound and the quantitation of related substances or degradant compounds. Solutions for these challenges include increasing the sample concentration and/or injection volume of the sample.
In addition, online procedures for concentrated sample preparations, UPLC, and/or detection, other than traditional UV/VIS, could be employed to meet the challenges. For dissolution testing, small-volume dissolution apparatus may be used and more efficient testing methods may be developed. “Use of techniques such as these has allowed Xcelience to provide analytical support of low-dosage form development,” says Dr. Chang.
For the fastest approach to get an active in First in Human clinical studies, Xcelience uses Xcelodose technology, which can fill active pharmaceutical ingredients directly into capsule shells to provide doses in the microgram levels. In addition, low-dose capsules or low-dose tablets can be developed.
To view this issue and all back issues online, please visit www.drug-dev.com.
Total Page Views: 9215












