



Issue:March 2016
NEXT-GENERATION SEQUENCING - NGS: Uniformity Across the Block
INTRODUCTION
Next-generation sequencing (NGS) allows researchers working across diverse life science disciplines to study biological systems with unprecedented throughput, scalability, and speed. From clinical diagnostics and forensics through to environmental science and now drug delivery, the application of NGS delivers a depth of genomics data that far exceeds conventional DNA sequencing techniques. However, NGS remains a high-cost pursuit. The risk of a single failed NGS run, for example, can cost in excess of $3,000. Securing complete precision and confidence at each step of the NGS workflow is therefore a critical pursuit during method development.
Quantitative Polymerase Chain Reaction (qPCR) technology is a core technique within NGS, employed for the robust quantification of library target molecules. It is essential that qPCR instrumentation deliver precise data at this point, or users risk ruining the NGS run before it even gets started. Unfortunately, many traditional qPCR systems suffer from imprecise temperature control, uniformity, and light bleed-through, which compromise the reliability of each run.
One way manufacturers are responding to these inefficiencies is to design qPCR technology that delivers complete thermal and optical uniformity across the entire sample block. This helps to guarantee that each run is completed reliably and accurately, mitigating the risk of costly NGS failure. This article explores how innovations in instruments, such as the Eco 48 real-time qPCR system (PCRmax), deliver exceptional uniformity, actively improving the reliability and productivity of the working laboratory.
NGS FOR DRUG DEVELOPMENT
Throughout the past 30 years, the development and maturation of high throughput NGS technology has transformed practically every facet of biological science. Within the clinical environment, NGS has become essential for assay development in disease detection and diagnosis. More recently, NGS has been deployed for drug development, particularly in the emerging field of personalized medicine.
NGS technology enables users to simultaneously process millions of parallel DNA sequences many orders of magnitude faster than traditional sequencing. Within drug development, NGS is increasingly used to rapidly generate genetic information to better understand the process of disease development and how patient-specific characteristics influence the response to new therapeutic treatments. NGS tests are increasingly employed across multiple disease areas, with particular success in the field of oncology. Comprehensive analysis of a tumor’s genetic profile enables researchers to study the development of a cancerous growth, its progression, and the emergence of drug resistance. In this way, NGS helps users produce pharmaceuticals with greater efficacy and performance and allows physicians to ensure patients are offered the most effective treatment available.
Today, NGS is a core technique throughout genomics research. However, despite advances in accessibility and usability, NGS remains a high-cost process. Initial capital expenditure in NGS
equipment ranges from $150,000 upward to $1 million. Moreover, the cost of driving these processes is equally high. Optimizing the conditions of each run to mitigate failure is therefore an essential aspect of method development.
Target library quantitation is an area in which cost-effective technologies, such as quantitative Polymerase Chain Reaction (qPCR), are available to streamline and stabilise the NGS process, maximizing returns from each run.
SAVING TIME & MONEY WITH QPCR-DRIVEN LIBRARY QUANTITATION
Target library quantitation is an essential step in NGS preparation, which often determines the success of the entire run. During library quantitation, fragments of target molecules are fused with adapters, followed by amplification and sequencing with polymerase chain reaction (PCR). The size of the target DNA fragments in the final library is a critical parameter for NGS library construction. If too little template is loaded onto the NGS platform, then the run will have low efficiencies. If too much is loaded, then the run not only risks low efficiency, but increased probability of complete failure. Robust and reliable library quantification is therefore critical.
Today, qPCR has become the technique of choice for NGS library quantitation, delivering precise analysis of strand amplification in real-time. During qPCR, dsDNA is amplified using primers with complementary sequences to the previously annealed NGS adapter. This results in a fluorescent signal being generated when the target strand is intercalated with DNA binding chemistry in the reaction mix. Capturing fluorescence in real-time enables precise quantification of amplification at appropriate stages of the reaction.
Real-time monitoring overcomes many of the inaccuracies associated with traditional end-point PCR, including unaccounted for variation in efficiency between samples and errors when extrapolating back to starting efficiency. Moreover, unlike end-point PCR, qPCR does not require a gel, eliminating much of the associated contamination risk. However, not all qPCR systems deliver the repeatability and accuracy required for secure NGS system loading.
Precise temperature control is fundamental to the success of the qPCR process. Block temperature defines key features of the reaction, including primer binding and the performance of the polymerase enzyme. A qPCR thermal system must ensure that all of the samples in the well are uniformly heated so they proceed through the reaction at an equal rate. However, standard qPCR systems only have a thermal accuracy of around ±0.5°C at the 50°C to 60°C range. This is often insufficient to precisely define the cluster density, and an NGS run may risk failure if the qPCR platform under- or over-represents the true library concentration. The importance of achieving this level of precision is highlighted by stringent MIQE (Minimum Information for Publication for Quantitative Real-Time PCR Experiments) guidelines for Qpcr experiments, which demands increasingly sensitive qPCR technology.
With the price of high-performance qPCR instruments being outweighed by the cost of failed or low efficiency runs, employing a robust quantitative qPCR technique that delivers precise heating uniformity is possibly the simplest and most cost-effective way to guarantee consistent NGS.
ACHIEVING UNIFORMITY ACROSS THE BLOCK
Traditional qPCR systems employ solid Peltier-heated thermal blocks to drive thermal cycling. These systems are designed to heat the entire block to a temperature exceeding that of the required reaction before equilibrating to a desired plateau.
This is an energy-intensive process and the time it takes for all wells within the block to reach this point results in long run times and severely impacts the efficiency of the entire NGS run. More significantly, this method of heating contributes to high thermal non-uniformity (TNU) values of ±0.5°C and poor thermal ramp-rates. The combination of imprecision and inefficiency delivered by traditional solid-block qPCR means such systems are poorly suited to high-performance applications.
Recent advances in block heating technology overcome these issues. One significant innovation is the development of precisely uniformed hollow silver block structures through which a conductive fluid is passed to heat the sample. A single Peltier device is used to heat and cool the fluid, which is circulated evenly across all the sample wells by opposing agitators. This simple solution delivers both robust thermal performance, with TNU values below ±0.1°C at 95°C, and reduced run times. Standard 40 cycle PCR protocols take about 40 minutes to complete on these systems, while a fully optimized process can complete in only 15 minutes.
Advances in fluorescence monitoring technology have also improved the performance of qPCR techniques. High-performance optical systems enable real-time detection of up to four targets in a single reaction, and advanced detector arrays monitor the fluorescence from all wells, allowing the system to record every well, filter, and cycle without missing a single data point. Finally, the use of stable light emitting diodes (LEDs) contributes to accurate data generation and increases instrument longevity, decreasing depreciation costs. In combination, these innovations allow qPCR practitioners to meet and surpass all required MIQE guidelines, with no compromise to efficiency, cost, or speed. The following case study illustrates how improvements in thermal uniformity across the block deliver the sensitivity, precision, and overall efficiency required for high-performance NGS applications.
CASE STUDY: IMPROVING THE PRECISION OF QPCR
Forty-eight replicate samples were subjected to a High Resolution Melt (HRM) protocol using the PCRmax Eco 48. HRM enables precise analysis of genetic variations, such as quantifying single nucleotide polymorphs (SNPs), and is one of the most thermally demanding protocols in terms of accuracy.
Each of the 48 sample wells were filled with 1×108 copies of the starting template (100bp template based on Lambda phage DNA) in a 10-microliter final volume. The plate was sealed and centrifuged for 1 minute at 12000 rpm, and the un-optimized 40-cycle PCR protocol was performed in 43 minutes total. The template was amplified for 40 cycles (95°C, 10 secs; 60°C, 30 secs) using the GoTaq® QPCR Master Mix (2x) from Promega (part code A6001). Fluorescence data was collected at the end of the 60°C step. The results were analyzed using Eco study software to determine the quantitation cycle (Cq), the point at which fluorescence can be detected, and the melting temperature (Tm) values for each of the 48 replicates.
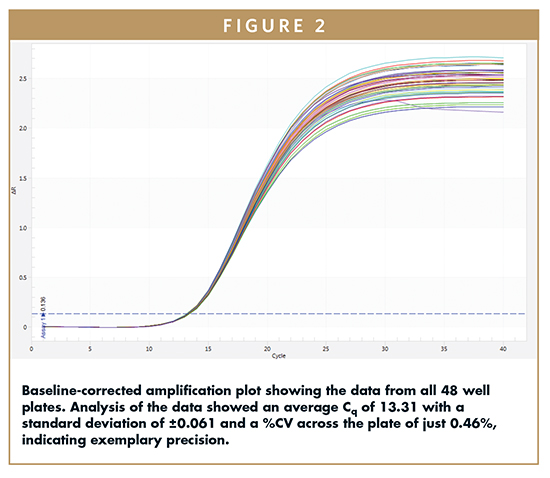
Figure 2 shows the baseline-corrected amplification plot for all 48 wells. The graph clearly demonstrates precision of amplification across the entire plate. Analysis of the data showed an average Cq of 13.31 with a standard deviation of ±0.061. This equates to a coefficient of variation (%CV) across the plate of just 0.46%, indicating exemplary precision.
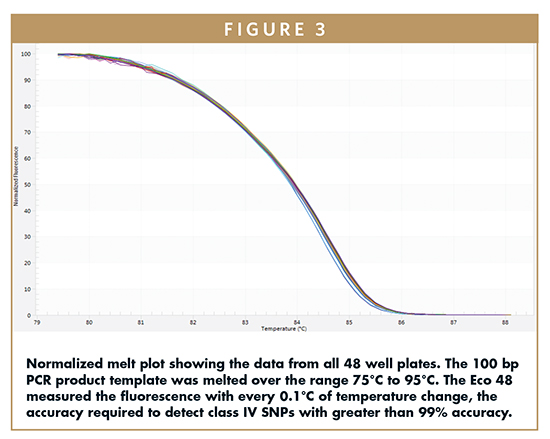
Tm is determined by running a melting stage following the amplification of the PCR product and is one of the most effective measures of block uniformity. The amplified product melted in the 75°C to 95°C range, and the Eco 48 measured the fluorescence with every 0.1°C temperature change, the accuracy required to detect class IV SNPs with greater than 99% accuracy. Figure 3 shows the normalized melt curve.
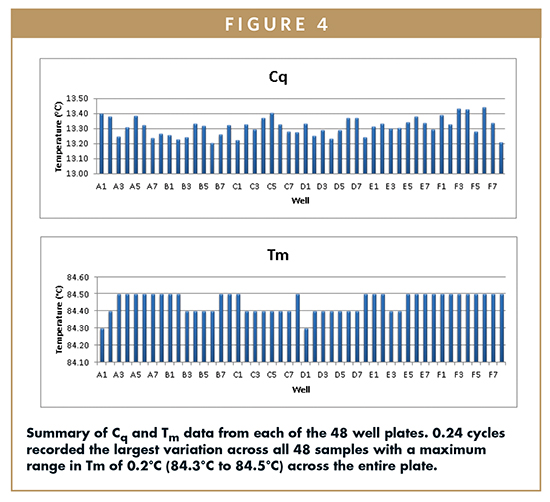
Tm average across all 48 well plates was recorded as 84.45°C with a standard deviation of ±0.058, equating to a %CV across the plate of just 0.07%. This suggests that excellent temperature uniformity is achieved across the block. Figure 4 summarizes the results for this experiment.
DRIVING THE FUTURE OF GENOMICS
Advances in NGS technology continue to drive development across the diverse genomics arena. Within drug development in particular, NGS-driven genetically targeted therapies are delivering improved treatment pathways and greater patient care for a range of diseases. NGS data now forms the bedrock for a range of applications, and the reliability of the run is integral to the productivity of this high-cost process. The increased confidence that greater thermal control provides during NGS has made qPCR an essential feature of routine sequencing. qPCR instruments with innovative block technology, such as the PCRmax Eco 48, deliver complete heating uniformity and rapid cycles in under 40 minutes, with the sensitivity to deliver ±0.1°C uniformity across the whole block instantly after every temperature change. These leaps in efficiency allow users to achieve consistent NGS runs, helping to further their critical genomics research.
To view this issue and all back issues online, please visit www.drug-dev.com.

Dr. Andrew Birnie is an experienced molecular biology specialist. He currently works as Business Development Specialist at PCRmax (a Bibby Scientific Company); specializing in applications, technical support, and marketing. His expertise and interest in molecular biology allow him to offer significant input into product development. Prior to joining PCRmax, Dr. Birnie was a Molecular Biology Product Manager at Labtech Int. with his role involving the management of the Molecular Biology portfolio. Dr. Birnie earned his PhD in Veterinary Parasitology at The University of Glasgow, where his research focused upon the nematode caenorhabditis elegans, as a model organism for nematode parasites.
Total Page Views: 3155












