



Issue:April 2018
FDA UPDATE - The FDA's New Drug Approval Process: Development & Premarket Applications
INTRODUCTION
The Food and Drug Administration (FDA) is responsible for advancing the public health by helping to speed innovations that make medicines safer and more effective and by helping the public get the accurate, science-based information it needs to use medicines to maintain and improve public health. This publication emphasizes quality system approaches to the development and availability of new drug information presented in the proposed labeling of the product. In 2004, the FDA provided a guidance document for innovations, challenges, and solutions for new drug products that examine the critical path needed to bring therapeutic products to completion, and how the FDA can collaborate in the process, from laboratory to production to end use, to make medical breakthroughs available to those in need as quickly as possible.
DRUG DEVELOPMENT RESEARCH
One of the primary functions of a firm’s research project team is to coordinate the various studies necessary for the successful development of a drug candidate and to plan a timeline for developmental activities for its premarket application. This coordination is usually accomplished by preparing a detailed drug development plan and monitoring the research process. This requires analyzing the information and studies as they relate to the proposed drug candidate type commonly referred to as a novel chemical entity for disease indication and the intended use (ie, cardiovascular, cancer, CNS indications, diabetes, etc). This includes types and duration of therapies (ie, acute or chronic situations with one or a few doses adequate for treatment modes). It’s also important to consider routes of administration (ie, intravenous or infusion ornonintravenous such as oral, pulmonary, subcutaneous, intramuscular, dermal, etc). The timelines for the various studies and their integration into a formal drug development plan are compound-specific and dependent on the availability of resources within the various departments of the sponsor firm and approval of CROs. At the same time, the designation of pertinent milestone events and the critical path are compound-specific and firm-specific. Additionally, studies, such as bioavailability for a candidate drug may be necessary. Other studies, such as potency, immunogenicity, and toxicity may be required. The formation of various project teams requires coordination for the successful development of a drug candidate for premarket applications submitted to the FDA.
STUDY DESIGN
The research teams should carefully review and evaluate the prototype design studies for the candidate drug as to how it is similar or different from the intended clinical use and determine whether the appropriate subject population and resources are available at a given institution and whether any requirements unique to the protocol can be met at that site. In addition to identifying the type of study, the team should consider as to how the protocol requirements compare to the routine standard of care for the selected patient population. The team should consider as to how the drug dosing will be determined.
PRECLINICAL DRUG DEVELOPMENT
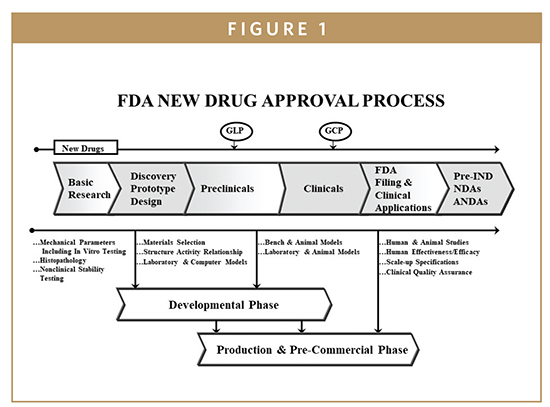
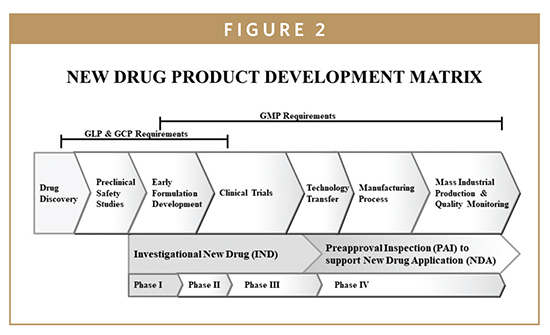
The drug candidate is subjected to a number of preclinical studies to establish and characterize its safety profile. New drugs must be shown to be safe and effective in human subjects before FDA approval. The drug company must first convince the FDA that the drug is reasonably safe to use in humans to evaluate safety and efficacy in clinical studies. This is established through preclinical laboratory testing, including testing in animals. These studies of a new compound or drug, generally performed in animals, are referred to as “preclinical studies” (Figures 1 & 2). Preclinical studies help establish boundaries for the safe use of the treatment when human testing or “clinical trials” begin. The sponsor of the new drug product submits an IND application to the FDA requesting permission to initiate clinical trials. The results from preclinical studies are documented in scientific publications or technical reports and used to prepare as part of premarket submission for the initiation of human clinical trials. The preclinical studies on a potential drug substance are required to follow Good Laboratory Practices (GLPs) regulations. GLPs govern laboratory facilities, personnel, equipment, and operations. Compliance with GLPs requires procedures and documentation of training, study schedules, processes, and status reports, which are submitted to facility management and included in the final study report to the FDA. The preclinical studies data are gathered to reach the goal of potential therapeutic effect and reasonable safety index and the drug sponsor must notify the FDA of its intent to test the potential new drug in humans. The application to request permission to begin human testing is commonly referred to as an Investigational New Drug (IND) application. The IND allows the use of an investigational drug in human subjects for the sole purpose of conducting clinical trials.
GOOD LABORATORY PRACTICE (GLP)
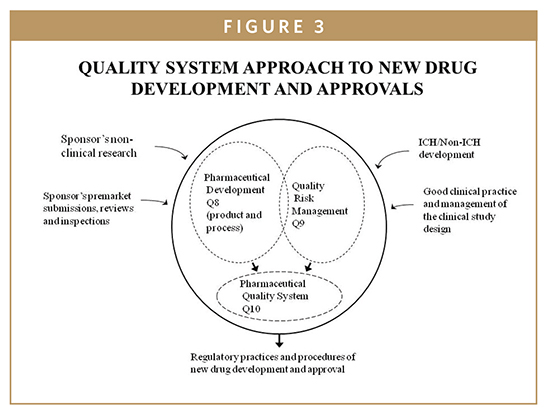
GLPs are the regulations for the nonclinical laboratory studies to support INDs). FDA regulations applicable to GLPs are provided in (21 CFR, Part 58). GLP regulations require protocols for standard methods, facilities, equipment, test controls, records and reports, audits, and inspections to be used in conducting preclinical and nonclinical laboratory studies that are used to ensure the quality and integrity of data provided in INDs. Nonclinical studies include in vitro and in vivo experiments for the new drug safety profiles. GLP standards relate to both the design and the conduct of laboratory studies and the qualifications of the personnel and facilities involved with the experiments. The purpose of GLP is to ensure the integrity of the nonclinical safety data, such that an evaluation of the study quality and inter pretation of the study results may be done with confidence. Guidance documents related to GLPs are issued by the FDA and ICH (International Conference on Harmonization) as illustrated as part of Quality System Model presented in Figure 3. The GLP highlights are:
-SOPs written for routine or standard practices in the laboratory
-Personnel involved with the studies are trained and experienced
-The facilities are appropriately designed and maintained
-A group, commonly called quality assurance or QA, monitors and checks the results from the studies to ensure that the experiments are conducted in compliance with regulations
GOOD CLINICAL PRACTICE (GCP)
FDA regulations applicable to GCPs are provided in (21 CFR 312). The FDA has published a consolidated guideline of GCP in conjunction with the ICH guideline {E6, 62 Fed. Reg. 25692 (1998)}. The consolidated guideline for GCP is intended to provide a unified standard for conducting clinical studies. These standards apply to all aspects of clinical trials, from protocol design, monitoring, and auditing, to recording, analysis, and reporting of clinical data presented in new drug applications to the FDA. Guidance documents related to GCPs are issued by FDA and ICH as illustrated as part of Quality System Model presented in Figure 3. The overall aim of GCP is to protect public health and the rights, welfare, and confidentiality of study participants. The GCP process is intended to ensure that all data and reported results are credible, accurate, and evidence-based. While GCP places emphasis on the clinical accuracy of results, it also deals with the importance of the processes used to conduct clinical trials.1-5 FDA is focused on the conduct of clinical trials and embracing GCPs as a “Quality System Approach to New Drug Development and Approvals”- (Figure 3). According to this approach GCP refers to the collection of regulations and requirements that must be complied with while conducting clinical trials. These regulations apply to manufacturers, sponsors, clinical investigators, and institutional review boards.1,5
PRINCIPLES & PROCEDURES FOR NEW DRUG APPLICATIONS
The FDA new drug approval process begins with research plans involving basic research, laboratory, and animal testing. This initial stage includes discovery and development of prototypes involving preclinical and clinical studies of new drug materials to be reviewed and approved by an institutional review board (IRB). These IRBs exist in hospitals, university medical centers, and private clinical research institutions at which clinical trials take place. Before a clinical trial is initiated, foreseeable risks are weighed against the anticipated benefits for the individual trial subject and the intended clinical population. Generally, a clinical trial is initiated and continued only if the anticipated benefits are feasible (Figure 1). The FDA filing and premarket applications consist of the following categories:
1. Investigational New Drug Application (IND)
2. New Drug Application (NDA)
3. Abbreviated New Drug Application (ANDA)
For a drug manufacturer to introduce a product in the market for human use, a multiphase procedure is followed. This procedure begins with a number of preclinical or “prior to human” testing, followed usually by three phases of human studies.1,3,5 New drugs are also subject to a fourth phase, known as post-market surveillance, which may require additional trial data. The FDA has published detailed information on the drug development process (www.fda.gov/cder/handbook/develop.htm). This publication not only addresses the importance of interactions between the sponsor and the FDA, but also emphasizes the interactions between the various stages of investigational studies and the continuing dialogue with the FDA review status throughout the development and completion of premarket application.
IND is a submission to the FDA requesting permission to initiate a clinical study of a new drug product in the US. The main purpose of an IND is to seek an “exemption” from the Act’s prohibition of introducing any new drug into interstate commerce without an approved application, or to allow a firm to request permission to ship an “unapproved drug” or import the new drug from a foreign country. IND allows a company to initiate and conduct clinical studies of their investigational drug product. These studies are used to gather significant evidence of reasonable safety and efficacy data about the candidate drug compound in humans. Numerous meetings between the sponsor and the FDA take place during these studies. The requirements for the format and content of the IND application are provided in (21 CFR Part 312).
NDA is a premarket submission to the US FDA requesting to obtain approval for marketing a new drug in the US. The FDA reviews the NDA application and ultimately makes the decision on whether the drug application is fillable (Figure 1). Prior to making the decision, the FDA will arrange for an advisory committee meeting of outside experts to seek their recommendation in regard to the approvability of the premarket application. The recommendations of an advisory committee are not binding, but the agency considers them very carefully when making approval decisions. The NDA submission is organized into specific technical sections, which are evaluated by specialized FDA review teams. The review teams recommend approval or disapproval. The FDA authority to require an NDA (prior to marketing the drug product in the US) is drawn from section 505 of the Food, Drug and Cosmetic Act {21 USC 355}. The content and format of an NDA is laid out in 21 CFR Part 314 and guidance documents published by FDA (http://www.fda/cder/guidance/5445fnl.htm#Toc77574464). When all the aforementioned steps are completed, the FDA inspects the manufacturing plant to ensure the sponsor’s facilities are capable of manufacturing the drug in compliance with the FDA’s current good manufacturing practices (cGMP) regulations (Figure 2).
ANDA is for new drugs approved which must be pharmaceutically equivalent and bioequivalent to predicate product, usually an innovator or pioneer drug (reference listed product 21CFR 314.94) ANDA is a submission to FDA as an ANDA .These applications are called “abbreviated” because the generic drug manufacturers are not required to include preclinical or clinical data to establish safety and effectiveness because those characteristics were already established by the manufacturer of the innovator drug through the NDA process. The sponsor of an ANDA must provide information and data demonstrating that the drug product is bioequivalent to the innovator drug and the proposed use and labeling is identical to that of the reference innovator drug. ANDA sponsor manufacturers are subject to the same inspection requirements that apply to manufacturers of new innovator drugs.
CLINICAL TRIALS
Clinical trials are an integral part of new drug discovery and development; and they require review and evaluation by the FDA before the new drug product can be brought to market. Before submitting an NDA, the sponsor must conduct preclinical and clinical studies designed to demonstrate the safety and efficacy of the drug product. Clinical trials involve studies of human subjects where the protocol-designed studies provide information and data to support the NDA submission to FDA. Clinical trials may be classified by their stage and phase in the product life cycle and are generally categorized into three phases (Figure 2).1-5 Clinical trials require careful planning and consideration of the types of subjects to be enrolled. The main purpose of clinical trials design objectives is to test a hypothesis and ultimately to reach a conclusion as to whether a drug product has any effect on the human body and the disease condition in which it is being tested. Additionally, the drug product improves the subject’s health or quality of life, have an advantage over the current treatment available for that disease or condition, and can be administered safely to that subject. Sponsors of drug product studies are required to control risks to clinical trial participants. It is critical that all personnel involved in clinical trials understand the regulations and guidelines that govern the protection of human subjects while evaluating the efficacy of the products.
CLINICAL TRIAL PHASES
Clinical trials for new drugs typically consist of three phases. Phase I involves a relatively small number of subjects (less than 100) intended to gather initial safety information. Its purpose is to determine a safe dose range in which the drug can be administered, metabolized, and pharmacologically effective with minimum toxicity. The safety and pharmacokinetics of the doses in these studies usually include testing to help establish the relationship between drug dose and plasma concentration levels, as well as therapeutic or toxic effects. The results of the Phase I studies are used to develop Phase II.
Phase II involves a large number of subjects who have the disease or condition the drug product is intended to treat (usually 100-300). The purpose of Phase II studies is to determine a minimum and maximum effective dose (dose-ranging study and pharmacokinetic data). Clear evidence is established to confirm that the mechanism of action observed in animals is observed in humans. Phase II may be divided into two subparts: Phase IIa is a pilot study, which is used to determine initial efficacy, and Phase IIb uses controlled studies on several hundred patients. Sufficient data regarding tolerability and efficacy of a number of different dose regimens should be available to support the dose regimen to be evaluated in Phase III trials. At this point, the sponsor and the FDA usually confer to discuss the data and plans for Phase III.
Phase III studies are considered “pivotal”, designed to collect all of the essential data to fulfill the safety and efficacy criteria that the FDA requires to approve the new drug application for the US marketplace. Phase III studies are usually very large, consisting of thousands of patients usually in double-blind, randomized, controlled studies that are often conducted at multiple sites. In this phase, detailed data are gathered about the effectiveness of new drug compound in comparison to control treatments. Subjects are followed to evaluate side effects and safety. Additionally, Phase III studies establish effectiveness of final formulation, indications for clinical use, labeling, marketing claims, drug product stability, packaging, and storage conditions (Figures 1 & 2). Upon completion of Phase III, all clinical studies are completed and the sponsor submits an NDA to the FDA for premarket approval to market the new drug in US.
FDA’S GOOD MANUFACTURING PRACTICES (CGMPS) & PREAPPROVAL INSPECTIONS (PAIS)
Current Good Manufacturing Practices (cGMPs)
Pharmaceutical cGMPs (Title 21 CFR 210 & 211) are the part of quality assurance practices that ensure the drug products are consistently produced and controlled in conformance with quality standards (Figure 4).1 They are known as current manufacturing practices, processing, packing, or holding of drugs and current manufacturing practices for finished pharmaceuticals. The ICH Q10 was adopted by US in 2009. The FDA guidance, “Quality Systems Approach to Pharmaceutical CGMPs” describes the aim of the agency to help manufacturers implementing modern quality systems and risk management tools to meet the requirements of the agency’s current approaches to cGMPs. The implementation of ICH Q10 throughout the product life cycle facilitates and strengthens the link between drug development and manufacturing activities. In addition to ICH Q10, the FDA adopted industry sponsored guidelines for continuous quality improvement (ISBN 0273 -3099). The FDA appears committed to support ways to promote drug development, and is willing to accommodate NDA sponsors to use improved quality management approaches to foster innovations and improvements. These approaches help enhance the consistency and coordination of the FDA’s drug quality regulatory programs, in part, by further integrating enhanced quality systems approaches into the agency’s regulatory processes concerning review and inspection activities. In reference to NDAs, cGMPs include quality system approaches whereby the sponsor addresses the specifications of the drug product and the manufacturing process controls from the prototype design to the production and release of the finished product (Figure 4). The FDA’s CGMP regulations do not prescribe in detail how a manufacturer must proceed as it designs and manufactures a specific drug product. Instead, a framework is presented requiring the manufacturer to develop and follow procedures and to fill in the appropriate details for a particular drug; however, the most important point behind GMP regulations is that quality must be designed and built into a product. As development proceeds, the active drug substance and dose form must be manufactured at larger scales. This scale-up may introduce variations in the manufacturing steps of drug product. Thus, it is critical to monitor the drug substance and product for variations during development and manufacturing processes. The emphasis of design controls drug GMPs should be on products that conform to defined user needs and intended uses. For NDA applications, it is essential to have data showing that the product and active drug substance have documented stability in the packaging that will be used for marketed product. FDA GMP regulations require information about all the steps of the manufacturing process from incoming materials to final distribution of the product (Figure 4).

Drug Product Life Cycle (DPLC)
The design phase of drug product life cycle is the most important development stage in regard to the lifecycle of the drug. It is at the design stage that the inherent safety and efficacy of a drug are established. The review and periodic management of design and processes involved in the drug product development are essential to maintain the drug quality toward completion of production specifications. Design maintenance activities during the development process ensures that design outputs are verified as suitable for manufacturing before becoming final production specifications.2 The flow diagram for GMP inspections represents the process flow for the FDA’s inspections for design control requirements. The FDA investigator verifies that the formulation, manufacturing, or processing methods are consistent with descriptions contained in the section of the NDA application. Manufacturing process flowcharts provide road maps to FDA investigators. They provide a detailed view of the process, and increase understanding of how the process flows. With a process flowchart, FDA investigator can identify critical control points of manufacturing processes.1-3
Hazard Analysis & Risk Assessment
The DPLC for an NDA drug product is an integrated development and marketing framework. The DPLC can be divided into the following segments:
-Early product cycle (concept, prototype)
-Mid product cycle (pre-clinical, clinical, manufacturing)
-Late product cycle (marketing, commercial use, continuous quality improvement, and design controls)
All segments of DPLC are interconnected to every other phase with the final drug product providing built-in quality and process improvement. Issues learned from one part of a life cycle are applied to the development of the next generation. The essential principles of DPLC are composed of management responsibilities, quality assurance, and drug design monitoring units (Figure 5). The ICH quality system approach requires sponsors of NDAs to establish and maintain procedures to control the design of the drug product in order to ensure that specified requirements are met. As previously mentioned, intrinsic quality of the NDA drug product including its safety and efficacy are established during the design phase. Thus, appropriate drug design controls are observed and maintained during production stages of development so that finished drug products are safe and effective for their intended clinical use and points of disposals. Process validation (PV) is a requirement of the FDA’s cGMP regulation and typically, the drug industry approach to PV has been to evaluate prospective batches incorporating risk analysis in regard to complexity of the manufacturing process or dosage form, unit operations, or critical control points in developmental stages.1
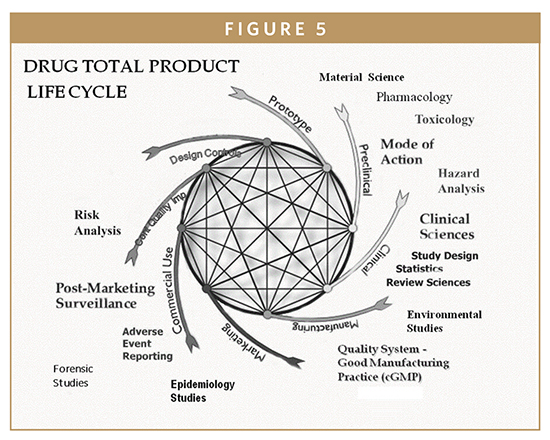
A quality system approach to new drug development and approval starts by defining the intended use, indications for use, drug design controls, impact of risk analysis, and any foreseeable drug errors and clinically incorrect patient diagnosis and/or treatments (ie, adverse events). The FDA’s 21st century cGMP and ICH initiatives (such as Q8 Pharmaceutical Development, Q9 Quality Risk Management, and Q10 Pharmaceutical Quality System), evolved into new regulatory practices and procedures for NDA applications and approvals (Figures 3 & 5). Risk management can be applicable in several areas of PV, from early process/development through maintenance of validated stages during manufacturing processes.1-5 Some of the benefits of science- and risk-based approaches during PV are as follows:
-Benefits process understanding by proactive identification of failure modes (hazards), and managing the identified risks as early on in the product life cycle
-Enables that high risk, critical aspects of the process are well recognized by appropriately designed studies
-Monitoring of risks reduces product and process failures
It is important to assess the risks in each manufacturing process steps. The assessment starts by identifying the potential risks then controlling them to an acceptable level to ensure that drug product consistently meets approved quality standards.1 The FDA’s Quality by Design (QbD) guidances provide a sound framework for design controls from product development to the commercial manufacturing processes and for post-development changes and optimization. The QbD concepts are outlined in ICH Q8, Q9, and Q10 guidelines. These ICH documents are already adopted by the FDA. The QbD approach can be maintained throughout the life cycle of the product to facilitate continuous quality improvement (CQI). In contrast, previously, traditional pharmaceutical manufacturing relied heavily on end product testing, and the process typically lacked the flexibility needed to respond to variables encountered during manufacturing processes. The application of Hazard Analysis Critical Control Points (HACCP) principles identifies critical control points (CCPs) in the manufacturing process that require control monitoring because of detection of out-of-limits or drifts when they occur.1-5 The HACCP system provides a focus on the CCPs most likely to control product safety. This approach allows FDA reviewers and investigators to evaluate CCPs over time by examining a firm’s monitoring and corrective action records. Investigators can verify the HACCP application by confirming that significant product safety hazards are properly identified and the appropriate controls are in place.
SUMMARY
New drug applications are reviewed primarily for safety and efficacy with regard to their claims for intended clinical use. The FDA’s mission is to facilitate the development of the premarket review and evaluation of INDs and NDAs. A central theme over the past few years has been a standardized approach to evidence-based review and evaluation. The FDA emphasizes the Quality System approach to design of studies by providing oversight and objective review by setting thresholds for product safety and effectiveness by ensuring that organized data and appropriate labeling are present in support of the new drug’s intended and clinical use.
REFERENCES
1. Aziz, K.J. FDA’s Design Control Requirements for Biomarkers in Drug Development. J of Drug Development & Delivery. 2017:3:1-6.
2. Aziz, K.J. et. al. Do it by Design: An Introduction to Human Factors in Medical Devices 1996. (http:www.fda.gov/cdrh/humfac/doit.html).
3. Aziz, K.J. The FDA’s Perspective on Clinical Devices: Premarket Review and Evaluation Process. J Biomed Lab Sci. 1993,5:151-158.
4. Aziz, K.J. Global Harmonization and Quality System Applications for Clinical Diagnostics. Clin Lab Med. 2001:39:76-78.
5. Aziz,K.J. Clinical Trial Design: Integrating Biomarkers into Drug Development. Clin Lab and Analytical Sciences. 2007:30:10-19.
To view this issue and all back issues online, please visit www.drug-dev.com.

Dr. Kaiser J. Aziz is the former Director of Mechanics and Materials Science and Associate Director of Clinical Devices for the FDA. He is currently an independent consultant for clinical research, product development, and training. He has extensive regulatory experience in medical devices and pharmaceutical premarket evaluations and approvals. He has served as an adjunct faculty in the Department of Medicine and Physiology, NIH, Graduate School, where he developed and taught courses and workshops in Applied Clinical Trials. He has been a frequent invited speaker and educator at the Center for Health Sciences, Virginia Polytechnic Institute and State University, where he developed and taught medical device and pharmaceutical risk management courses and workshops. His expertise includes FDA’s Quality System Inspection Technique (QSIT) and medical products risk management using hazard analysis and critical control points (HACCP) Applications. Dr. Aziz earned his MS from Michigan State University, his PhD from American University, and a Post Doctorate in Health Services from the University of Southern California.
Total Page Views: 52756












