



Issue:April 2016
DRUG DEVELOPMENT - Don't Overlook Key Preclinical Research
INTRODUCTION
Critical factors impacting drug delivery, such as the overall pharmacokinetics profile (PK) and the pharmacodynamics profile (PD) of the compound, are often impacted by solubility, dissolution rate, and dissolution location of the drug once it is administered, especially for oral medications.
Animal-based toxicology studies are a pivotal part of any drug development program in which the drug is typically administered orally. The goal for any drug development team is to test and validate options that will provide optimal administration and uptake, and to identify dose-limiting toxicity levels.
However, in a rush to initiate such animal-based toxicity testing, many drug development teams skip key laboratory-based testing, simulation, and modeling options. This results in missed opportunities to thoroughly characterize the crucial solid-state materials properties that will impact the behavior and performance of the active pharmaceutical ingredient (API).
Because characteristics such as crystal form, size, surface area, and more (Figure 1) have a direct impact on the most appropriate and advantageous form of the API, it is in the drug discovery team’s best interest to partner with material scientists and formulation experts earlier in the process, to develop the needed insight earlier — before animal-based testing is initiated. Such an approach allows the team to quickly identify the most relevant solubility (kinetic and equilibrium), bioavailability, and toxicology values that are needed to accurately demonstrate the drug’s capabilities and initiate human-based clinical trials.

Too often, a trial-and-error approach is used, especially by smaller biopharmaceutical companies, which may lack the necessary internal resources or expertise (Figure 2). However, when disappointing early testing does not yield the anticipated results, the team invariably ends up going back to the drawing board, to carry out a series of iterative changes (for instance, altering the API phase or its polymorph, or varying the formulation) in an effort to achieve better test data. Such late changes typically require additional or different toxicity and efficacy studies to be carried out, which drives up costs and adds complexity and risk to the process.
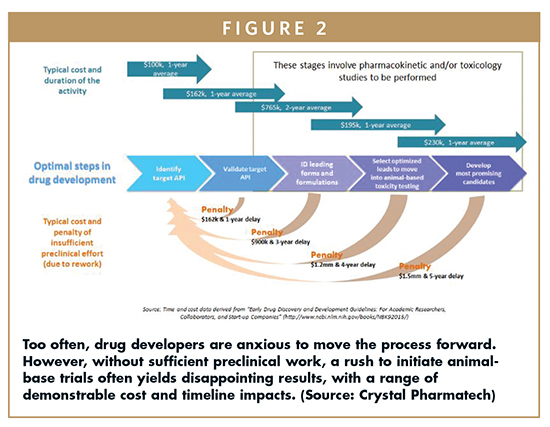
The following discusses an approach that can help the team develop greater depth of knowledge earlier in the process, and use it to realize shorter overall timelines, and reduced risk and cost. When smaller biotechnology companies do not have the required solid-state research capabilities in house, they should consider partnering with a contract research organization (CRO) that has broad and deep drug discovery and drug development knowledge and expertise, particularly with regard to solid-form selection and screening, toxicology formulation screening, excipient selection, and more.
FORM & FUNCTION
Most APIs can be produced in a variety of solid forms (ie, as a salt, a co-crystal, in free form, as a solvate or hydrate, in anhydrous free forms, in metastable forms, or as a polymorph, Figure 3). The specific API form may have a direct impact on animal-based toxicology testing results, and on other later drug development, scale-up, and production pathways.
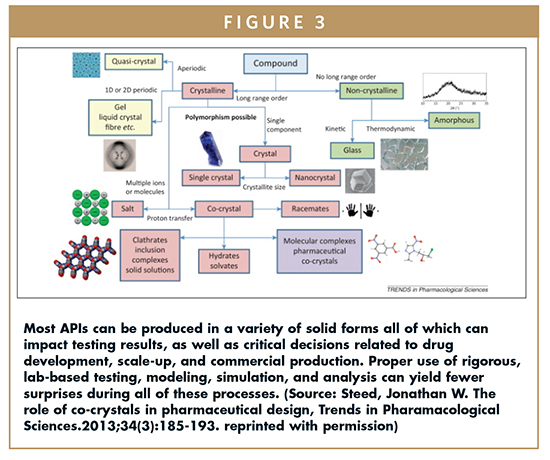
In most toxicology studies, the drug is administered as a solution or suspension. Nonetheless, it is the nature of the solid API itself that dictates the nature of the solution being prepared for testing (in terms of how much of the drug can be dissolved to reach the maximum dose, how much of the drug will stay in solution or remain in a suspension solution, and so on). For example, unwanted precipitation of the API would change the absorption, bioavailability, and pharmacokinetic characteristics of the candidate drug thereby significantly altering the preliminary testing data.
When rigorous lab-based testing, modeling, simulation, and analysis is used to better characterize the pharmacokinetic properties of the investigational drug, the team is able to more accurately predict and validate bioavailability and the concentration of the drug in the blood or urine over time during animal-based toxicity testing (that is, the PK profile). This helps the researchers to isolate and reduce avoidable variability earlier in the process, so that the actual testing data truly reflect the inherent variability of the compound itself, in terms of metabolism, excretion, and so on — not just variability that may be the attributable to too many other “moving parts.”
FOCUSING THE EFFORT EARLY
The behavior of the solubilized API will vary with the exact nature of its liquid-based formulation. To minimize the impact of such variation, the drug development team must carry out sufficient lab-based testing to accurately characterize the conditions at which the concentration of the API will change over time (for instance, precipitating out of solution), and to anticipate how the precise formulation will function in the gastrointestinal tract. To do this, lab-based screening techniques should be used to evaluate solubility/dissolution rates and possible precipitation of the competing candidate compounds in the face of simulated gastric fluids and simulated intestinal fluids.
Similarly, both conventional and non-conventional formulation options (for instance, using a spray-dried dispersion formulation) should be considered prior to the initiation of animal-based testing. The goal is to rule out sub-optimal options and hone in on the most-promising forms and formulations as the process moves forward into the toxicology-testing phase.
Ideally, early preclinical drug development efforts should incorporate the following types of solid-state research:
-Polymorph/salt/co-crystal screening
-Non-conventional formulation screening
-Thermodynamic form relationship
-Solid-form selection
-Single-crystal growth (to produce individual crystals that are acceptable for single crystal x-ray diffraction (SCXRD) structure determination)
Similarly, various options that are available to enhance the solubility of the API in solution should also be investigated. The goal is to develop a rational framework to do the right type of testing at the right time for the project at hand, in order to avoid missed opportunities and reduce excessive or greater detail later).
When material characteristics (and hence behavioral characteristics in the human body) are not thoroughly investigated and characterized early enough in the process, the initial animal-based toxicity testing can produce “muddy data.” Such data suggests confusing or disappointing results that don’t accurately reflect the actual capabilities of the API, but rather diverge greatly from what was expected on a dose-proportional basis. Such testing data may appear erratic, but in truth, the variability stems from the form or formulation being administered, and is not necessarily a reflection of the efficacy or toxicity of the API or compound itself.
If the investigational compound shows poor exposure during initial animal-based toxicology testing, the team will want to understand the mechanism that is to blame. Common factors to be considered include the following:
-Poor permeability/efflux, molecular weight, charge, other factors
-Fast metabolic degradation/firstpass
-Chemical or physical instability
-Poor solubility
-Inappropriate formulations (for instance, amorphous, nano, or cosolvent, or non-aqueous solutions)
-Particle size
-Crystal form
-Other
THE BUSINESS CASE
Any efforts to improve the preclinical development through better material science research will have direct and strategic business implications, with return on investment coming from the ability to:
-Shorten timelines (and therefore potentially shorten the time to market)
-Maximize earnings potential, revenue, profit margin, and market share (by receiving regulatory approval sooner)
-Minimize competition in some cases (through market exclusivity and other benefits if the product is first to market for a given indication)
-Eliminate unnecessary experiments and PK studies
-Reduce the number of animals required for toxicology studies
-Reduce overall risk associated with the program
And importantly, robust preclinical drug development efforts have the potential to strengthen the overall value of the drug portfolio for potential partnership or acquisition opportunities — from a due diligence perspective — by reducing risk, developing more-robust data, and potentially shortening the overall regulatory and commercialization pathway.
USE A RIGOROUS METHODOLOGY
The goal is to not just throw every tool at the problem, but rather to use a rational approach — using lab-based techniques (Figure 4), modeling, and analysis, well ahead of the onset of animal-based testing — to better understand the fundamentals of what is going on in your system. The following types of questions should be used to guide the effort:
-What is the ideal dose in solution?
-What is the ideal API form?
-Does particle size impact exposure?
-What is the ideal vehicle to use to produce the desired formulation?
-What happens when this solution encounters the anticipated pH that is representative of the conditions in the animal’s gastrointestinal system?
-Will the solution become gelatinous?
-Will the active ingredients come out of solution?
-Is the proposed solution able to meet the maximum required dose with no problems (physical, toxicological, or other)?
-Does the delivery vehicle itself present any safety concerns?
-Are preparation methods scalable?
-Can dose-limiting toxicity be predicted (and then verified during toxicity testing)?
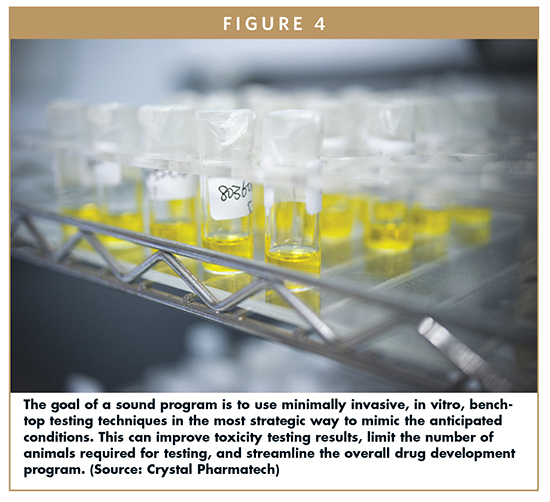
The ability to understand the basics early on — using minimally invasive, in vitro, bench-top testing techniques to mimic the anticipated conditions — can improve toxicity testing results, and importantly, can help to greatly limit the number of animals required for testing. The steps discussed briefly below provide a roadmap for carrying out pre-clinical, drug-screening and drug-formulation efforts:
1. Generate & Identify Candidate Compounds: characterize promising leads and develop small-scale scale-up routes
2. Phase Characterization: Evaluate API phase options and solubility profiles
3. Phase Evaluation: Evaluate stability and bioavailability of the candidate compounds
4. Phase Selection: Start making decisions about scale-up, formulation considerations and process control aspects
In an effort to save time and money, and start generating data, most of today’s drug development teams skip past Steps 2, 3, and 4, jumping right from Step 1 to the initiation of animal-based toxicology studies. As noted earlier, investments of time and effort made during Steps 2, 3, and 4 are well spent, providing the biggest possibility of payback over the long run.
ACKNOWLEDGEMENT
The authors would like to thank Suzanne Shelley of Precision Prose, Inc. (New York, NY; Email: SuzanneAShelley@yahoo.com; www.linkedin.com/in/suzanneshelley) for her assistance during the development of this article.
REFERENCES
1. Gu, Chong-Hui. Rational Formulation Design Through Biopharmaceutical Modeling, Solid-form Selection and Automated Formulation Screening, Webinar presentation (Accessed at: http://www.crystalpharmatech.com/resourcessh ow.asp).
2. Toussaint N, Bak A. Integrated Phase and Formulation Selection to Support GLP Toxicology and First-in-Human Studies (Accessed at: http://www.crystalpharmatech.com/resourcesshow.asp)
To view this issue and all back issues online, please visit www.drug-dev.com.

Dr. Robert Wenslow is Vice-President of Business Development at Crystal Pharmatech. He is a seasoned drug development scientist with a strong background in pharmaceutical characterization. He has extensive experience in all solid-state research issues relating to the pharmaceutical industry. Prior to founding Crystal Pharmatech, Dr. Wenslow spent 14 years at Merck & Co. His group at Merck supported, on average, 30 drug development projects per year. He also spent 2 years at Merck’s manufacturing site in Ballydine, Ireland, where he was responsible for creating and leading the first research-based Analytical Science Group in the Merck Manufacturing Network. Dr. Wenslow earned his PhD in Analytical Chemistry from Pennsylvania State University. He has presented papers at 10 professional conferences and published more than 25 peer-reviewed journal articles. He is also co-author on more than 10 patents. He can be reached at Robert_Wenslow@crystalpharmatech.com.

Dr. Ann Newman is Vice-President of Scientific Development and Head of the Scientific Advisory Board at Crystal Pharmatech, and pharmaceutical consultant at Seventh Street Development Group. She has more than 20 years of large pharma and contract research experience. For 10 years, Dr. Newman performed characterization studies on a wide range of pharmaceutical studies at Bristol-Myers Squibb, covering drug substance and product scale-up, from late drug discovery to launch and manufacturing. She has also held key roles at SSCI, Inc. and Aptuit, and holds an adjunct faculty in Industrial and Physical Pharmacy at Purdue University. She earned her PhD in Chemistry from the University of Connecticut, and is the author or collaborator on more than 45 publications, 90 technical presentations, and 55 webinars. She can be reached at ann@seventhstreetdev.com.
Total Page Views: 9551












