



Issue:May 2016
COMBINATION CORNER - How to Approach OTS Devices for Your Combination Product
In a recent survey1 of companies in the combination product space, individuals from Quality, Regulatory, and Research & Development were asked about the biggest challenges facing their firms as they strive to comply with the new FDA combination product regulations. One of the pain points they identified was related to confusion around how to handle combination product scenarios where one of the constituents is an off-the-shelf (OTS) medical device. This pain point is a broad statement; however, a myth that is consistently encountered is that OTS devices are already marketed and cleared medical devices by the FDA, and therefore no additional documentation is required. It’s perceived as a simple “plug-and-play” scenario. Unfortunately, it isn’t quite that simple.
Selecting an OTS medical device to use as a constituent significantly reduces risk and cost compared to developing a new device for the combination product (CP). On the other hand, it does not absolve the combination product manufacturer of the responsibility to show evidence that they have “developed” the correct device constituent. When thinking about device constituent development while using an OTS device, the literal translation of design and development does not apply because the OTS device has already been “designed and developed” and is commercially available. However, in this specific scenario, it applies to the design (selection) and development (qualification) of the device for use with the specific drug and/or biologic.
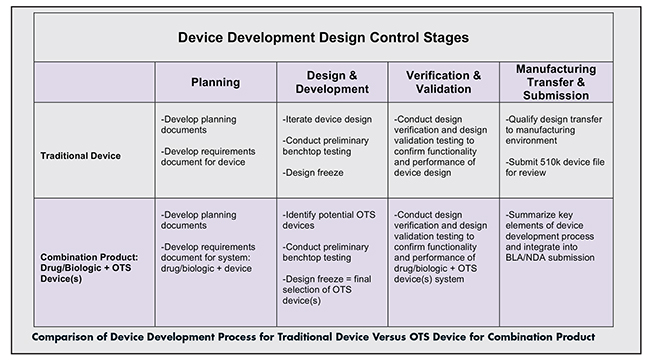
Confirming the selection and qualification of the OTS device for a CP should still follow the staged device design control process. The level of documentation that needs to be generated compared to a custom device development effort is significantly less, but the benefits of going through the process will yield a robust Design History File (DHF) that touches on the most critical elements of any device development process; including but not limited to requirements, selection of components, risk management activities, and verification and validation testing. Table 1 provides a high-level summary of the device development process (phases and key activities) for a traditional device development effort compared to a scenario where the combination product is utilizing OTS device(s). The following are three key points to think about when selected an OTS device for a CP, along with a mini-case study that demonstrates the benefits of taking an OTS device through the device design control process.
CONFIRM THE MARKETED OTS DEVICES HAVE AN INDICATION FOR USE THAT ALIGNS WITH INTENDED USE
The OTS devices were developed and cleared or approved by the FDA for a specific intended use. The documentation that the manufacturer has on file supports their indications and use conditions. It may or may not be appropriate for the targeted drug and/or biologic, but confirming this is not the responsibility of the OTS device manufacturer. Clearly understanding the boundaries established by the OTS device manufacturer will allow the drug/biologic company to assess potential fit of the identified device.
Mini-Case Study
A company identified an OTS ambulatory pump that had the right flow rate range for the subcutaneous delivery of its proprietary drug product and allowed the patient to be mobile while receiving treatments. A win-win situation (at least on the surface), and the company was planning on moving ahead with this device for commercialization. However, while the device constituent was going through the device development (design control) process for use with the drug, one of the device requirements was that the device label not be contraindicated for the combination product intended use. In challenging this requirement, it was identified that the ambulatory pump was indicated for intravenous use only, and contra-indicated for subcutaneous use. Reading the fine print identified a big issue and required that the company identify another pump for delivery of its product. The product development process did exactly what it was intended to do – confirm the OTS device chosen met the requirements of the combination product before the product proceeded too far into development or regulatory filing. Had the company not identified the issue, it may have found itself in a situation where it was preparing for a product launch only to have the FDA inform it the information supporting the use of the device constituent is not complete. Should the FDA had missed this discrepancy, the company would have been promoting the device for off-label use.
TRUST BUT VERIFY ANY CLAIMS MADE BY THE OTS DEVICE MANUFACTURER REGARDING THE PERFORMANCE OF ITS PRODUCT
Device manufacturers will provide key data regarding their device performance; however, the test methods and use-case conditions utilized to support their performance claims are typically not provided unless a specific ASTM/ISO standard is cited. But even in this instance, it is difficult to know if their test methods were validated. This level of detail is not typically made available by device manufacturers. As such, when selecting the potential OTS components, one needs to trust the data values that are being provided by the device manufacturer, but take on the responsibility to confirm these performance results – especially in instances where the performance of the drug and/or biologic is heavily dependent on a specific set of device parameters.
Mini-Case Study
A company was in the process of developing the requirements for the delivery of its drug. One key requirement for the device system focused on understanding the total residual volume of the delivery system in order to confirm the patient would receive the required minimum drug dose. The company indicated that when reading the OTS device manufacturer literature and adding up the claimed residual volume values (in this instance, there were multiple devices including a syringe pump and administration set), the total residual volume was acceptable and posed no risk to the patient not receiving the required minimum dose. However, it was recommended that the some benchtop studies be conducted to check if the values obtained under their intended use conditions (ie, a specific volume at a specific flow rate) would align with the published literature. For one of the two device components, the results yielded lower residual volumes than published by the OTS device manufacturer, while the second component yielded residual volumes that were three times higher than published residual volume of 0.2 mL. For a drug that’s total delivered volume is low (less than 20 mL per treatment), the difference between 0.2 mL and 0.6 mL of volume not delivered to the patient is significant, not to mention the cost of the drug being left behind. In this instance, the company was able to adjust the fill volume in the drug vial to accommodate the drug loss in the delivery system, and also initiate activities to identify an alternate device constituent that could provide a lower total residual volume.
REMEMBER THE OTS DEVICE(S) WERE NOT DEVELOPED WITH YOUR SPECIFIC DRUG/BIOLOGIC IN MIND
The OTS device(s) may have been developed as a “generic” device that will satisfy multiple needs and products. The drug/biologic company must understand the device company did not design the device with its specific drug or biologic, or use-case in mind. For example, a pump may present data on flow rate accuracy and/or residual volume; however, that data may have been collected using a limited number of types of liquids. Are those liquids representative of the intended viscosity and density of the intended drug/biologic product?
Mini-Case Study
A company was in the process of qualifying an ambulatory pump for the delivery of its biologic in the homecare setting. It should be noted that this specific biologic had a viscosity that was > 10 cp. A requirement identified for its product was the ambulatory pump could deliver a minimum of 20 infusions on a single battery. The OTS pump manufacturer published information that its device could function on a battery for over 30 hours, which collectively was greater than the time necessary to deliver the desired 20 infusions. This company had a design control process in place for devices utilized in a CP, and it proceeded to conduct testing on a number of different pumps to confirm that for its intended delivery conditions, the pumps could deliver a minimum requirement of 20 infusions on a single battery. During testing, it was identified that some of the pumps were not able to meet the minimum requirement of 20 infusions on a single battery. In addition, the pump-drive mechanism started to fail, highlighting a pump reliability issue. Conversations with the manufacturer yielded little value, and when testing evolved into understanding the weaknesses of the pump, the OTS device was immediately removed from consideration. The biologic company was starting to go down the path of understanding the design and reliability of the pump mechanism, which was not within the scope of its responsibility for the device and not its area of expertise. While the pump manufacturer indicated that its pump could handle a range of viscosities and flow rates, reality proved to be something different and highlighted that the manufacturer most likely had not done significant amounts of testing under more extreme use conditions.
SUMMARY
The aforementioned reminders seem so obvious, and yet, had these companies accepted information provided by the OTS manufacturer at face value and not developed and challenged their devices to the specific drug/biologic delivery requirements dictated by their product, they would not have uncovered the issues as early as they did. Discovering these issues early avoid the complications that would have surfaced had these OTS devices been released as part of the combination product. Utilizing OTS medical devices for drug/biologic combination products will continue to be a reality especially as devices like patch pumps and technology-integrated pen injectors are introduced. The bottom line is that while OTS devices are already “developed” and on the market, they should still go through the device development design control process from the perspective of the combination product, and then let the design control process determine if the device meets the specific requirements of the CP. Avoiding the design control process altogether for an OTS is NOT an option.
REFERENCE
1. EdgeOne Medical. Report: Highlighting Challenges in the New Combination Product Regulatory Landscape. EdgeOne Medical: Chicago, IL. February 2016.
To view this issue and all back issues online, please visit www.drug-dev.com.

Lilli Zakarija is Co-Founder and President of EdgeOne Medical Inc, an ISO 13485- certified medical device testing firm and consultancy focused on supporting combination products through the device development (design control) process. Prior to founding EdgeOne Medical, she developed and led the global device engineering function for Baxter’s BioScience division (now Baxalta) in support of all its combination (biologic and device) products and single-use, disposable medical devices. Ms. Zakarija earned both her BS in Biomedical Engineering, and Masters in Engineering Management from Northwestern University, and her Executive MBA from Kellogg School of Management.
Total Page Views: 2993












