



Issue:April 2014
ANTIBODY DRUG CONJUGATES - ADC Development Using SMARTag(TM) Technology
INTRODUCTION
Bioconjugates are an emerging class of biologics that combine the favorable properties of proteins, such as specificity and the ability to target distinct protein interfaces, with the advantages of synthetic small molecules, such as potency, bioavailability, and synthetic tractability. Early examples of bioconjugates include proteins linked to water-soluble polyethylene glycol (PEG), small molecule drug/protein combinations, and small molecule imaging agents attached to proteins. The body of published research focused on bioconjugation methods and applications continues to grow, and the number of bioconjugate compounds in development or on the market is steadily increasing. Although bioconjugates show great promise to improve existing therapeutics and create entirely new methods of treatment, there are also obstacles to overcome. The defining element of bioconjugates – the combination of large biologic and small synthetic molecules – is also its key weakness, on account of the inherent differences in the chemical properties of these components. Additionally, the chemical complexity of protein surfaces impedes the development of improved conjugation methods. Despite these challenges, there has been progress in advancing these complex compounds through clinical trials and successfully treating patients. These bioconjugate compounds include a subset of molecules known as antibody-drug conjugates (ADCs).1
ADCS: ADVANCED BIOCONJUGATES
ADCs are a class of bioconjugates that combine the target specificity of an antibody with the high potency of a synthetic small molecule. An ADC bioconjugate consists of a targeting component (a monoclonal antibody or mAb) that is chemically modified with a linker connected to a drug payload such as a cytotoxin (Figure 1A). The optimal ADC will leverage the innate characteristics of its distinct components to create a compound with an expanded therapeutic window, exhibiting increased potency with reduced toxicity and minimal side effects. After the antibody binds to its antigen on the target cell surface, the ADC is internalized by endocytosis. It eventually fuses with the lysosome and is degraded into its component parts, including the drug attached to a chemical linker. The active payload diffuses into the cytoplasm, where it induces cell death (or other desired activities) through mechanisms, such as tubulin destabilization and DNA alkylation (Figure 1B). ADC technology offers the promise of selectively reducing or eliminating tumors without affecting surrounding healthy tissue.2
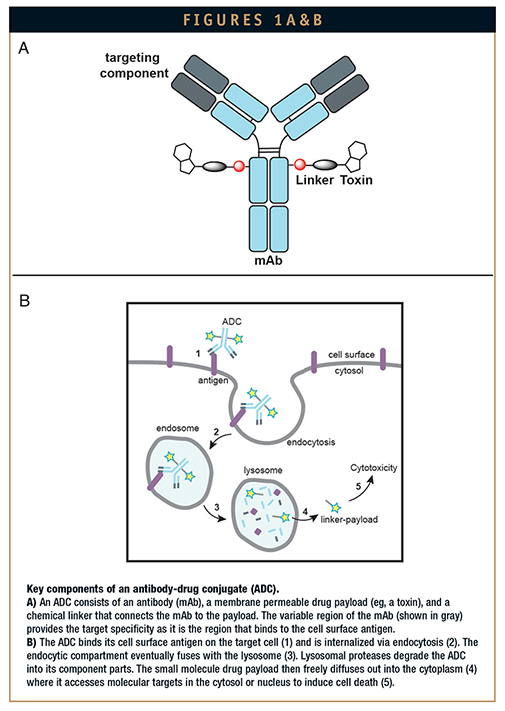
There are currently two ADCs on the market. Brentuximab vedotin (Adcetris®, Seattle Genetics) was approved by the FDA in 2011 for the treatment of Hodgkin’s and anaplastic large cell lymphoma. Adcetris is generated using maleimide chemistry to conjugate to cysteine (Cys) residues after mild reduction of the antibody. In 2013, trastuzumab emtansine (Kadcyla®, Genentech) gained FDA approval for the treatment of HER2+ breast cancer. The cytotoxic payload is conjugated to lysine (Lys) residues using a thioether linker system. Both Adcetris and Kadcyla have been successful at treating patients when other chemotherapies have failed. However, despite these successes, patients are still experiencing toxicity and significant side effects. The current ADC pipeline is large, with more than 100 ADCs currently in clinical trials; this effort underscores the industry’s belief that ADCs will be effective biotherapeutics. However, many challenges remain within the field of ADC technology (www.clinicaltrials.gov).
LIMITATIONS OF CURRENT ADC TECHNOLOGIES
The complications incurred with current ADCs are mainly a result of chemical promiscuity in the conjugate location on the protein peptide backbone. Traditionally, antibody-linker-drug conjugation chemistries have modified the thiol group of Cys residues, which can be targeted with maleimides, or the amino group of Lys residues, which can react with activated esters. Because antibodies contain multiple Cys and Lys residues, modifications using these conjugation methods are not site-specific. Therefore, placement of the payload cannot be precisely controlled. These random conjugation methods result in heterogeneous protein mixtures with variable drug-antibody ratios (DARs) and varied payload location on the protein peptide backbone. For example, with Cys conjugation, the reported DARs range from 0 to 8. With Lys conjugation, the location, or point of drug placement, is even more variable, with the resulting ADCs containing a statistical mixture of, potentially, hundreds of drug placement combinations.
The challenge with these methods is to ensure that product batches are reproducible from sequential production runs, considering the potential for ADC batches to have statistically varied drug payload placement. The ability to generate a consistent product is crucial for the regulatory approval of any drug. In addition, the analytics of such complex mixtures is tedious and costly. Furthermore, there are clinical ramifications caused by heterogeneous ADC mixtures. Any given ADC injection consists of a complex mixture of different chemical and therapeutically discrete active species. This heterogeneity creates significant therapeutic liabilities, with each distinct ADC having a different pharmacokinetic, efficacy, and safety profile. Conjugates with suboptimal toxin loads have reduced efficacy, while highly conjugated products are associated with increased toxicity. Additional complications are caused by the instability of the chemical linkages that are formed using conventional conjugation methods. For example, Cys-based maleimide bonds and Lys-based hydrazone bonds exhibit relatively short half-lives in vivo.3,4 The first ADC to gain FDA approval was gemtuzumab ozogamicin (Mylotarg, Wyeth/Pfizer), which used Lys conjugation to generate a heterogeneous ADC. This heterogeneity, along with an unstable acid-labile hydrazone linkage, contributed to significant toxicity in patients, which ultimately led to the drug being withdrawn from the market.2
SMARTAG TECHNOLOGY TO DEVELOP ADCS
One approach to solving the problem of ADC heterogeneity is to introduce chemistries that have tightly controlled reactivities in the context of a protein, resulting in site-specific conjugation. These bioconjugation reactions require bioorthogonal chemistries, or chemistries that are directed to a single chemical entity not found in naturally occurring polypeptides. To achieve chemical site-specificity, it is possible to incorporate a unique functional group, for example a novel side chain in an amino acid that has a distinct reactivity profile with respect to the 20 proteogenic amino acids. Several methods for the introduction of bioorthogonal reactive sites have been reported. These unconventional approaches include the introduction of exogenous Cys residues with enhanced reactivity, the incorporation of unnatural amino acids bearing synthetically derived side chains, and a number of chemoenzymatic approaches including the use of a microbial transglutaminase enzyme.1,5,6 Redwood Bioscience is pioneering an alternate chemoenzymatic solution to site-specificity, leveraging the naturally occurring formylglycine generating enzyme (FGE) to generate a unique, aldehyde-bearing amino acid residue known as formylglycine (FGly). Redwood Bioscience and Catalent Pharma Solutions have joined together to develop bioconjugates that incorporate Redwood Bioscience’s proprietary FGE/aldehyde tag technology and novel bioconjugation chemistry. This method, known as SMARTag technology, encompasses all the required components to produce best-in-class, site-specifically modified bioconjugates, including ADCs.
FGE is a naturally occurring enzyme endogenous to most prokaryotes and eukaryotes, including humans, that was discovered to be essential for the activation of a class of ubiquitous sulfatase enzymes that remove sulfate from biomolecules. Sulfatase activity requires an FGly residue, which is embedded in its active site. FGE is responsible for generating this FGly residue; it recognizes a consensus amino acid sequence, termed the sulfatase motif, and converts a Cys in the consensus sequence to an FGly through a novel oxidation process. The broadly defined minimal consensus motif recognized by FGE is the pentamer CxPxR, where x primarily consists of a neutral amino acid residue (Figure 2A). High conversion of Cys to FGly is possible when this small FGE target sequence, termed the aldehyde tag, is inserted into heterologous protein sequences and expressed using standard recombinant expression methods.7,8 The ability to take the aldehyde tag out of its natural context and place it into any desired protein with continued Cys to FGly conversion is the key element to SMARTag technology.
Recombinant proteins modified with the FGE aldehyde tag consensus sequence are produced in a wide variety of cell based expression systems including E. coli and mammalian cells.9 In mammalian cell lines, such as CHO, FGE is expressed in the endoplasmic reticulum (ER). The generation of FGly occurs co-translationally before protein folding and glycosylation is complete, obviating the need to express and purify a recombinant enzyme along with the target protein to be modified.10 In the context of producing high titers of recombinant protein, endogenous levels of FGE are often unable keep up with the high level of recombinant protein expression. However, FGE can be co-expressed with the aldehyde-tagged protein to ensure that Cys to FGly conversion goes to completion. Using standard cell culture techniques and cell line development to overexpress FGE, aldehyde-containing antibodies can be produced at high titers. Alternatively, conversion reactions can take place in vitro; purified aldehyde-tagged proteins can be incubated with purified recombinant FGE with robust Cys-to-FGly conversion. In vivo co-expression or in vitro incubation of FGE with an aldehyde-tagged protein yields products with complete conversion of Cys to FGly, which can be monitored and quantified using a mass spectrometry (MS)-based assay.11
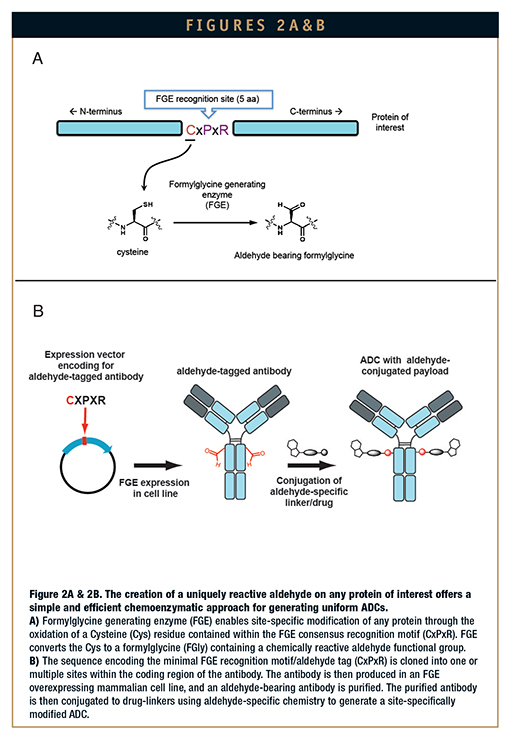
The SMARTag technology platform offers a simple and efficient bioconjugation method to produce homogenous, enhanced biotherapeutics. Because the aldehyde tag is a short, six amino acid sequence, it can be genetically encoded into any heterologous protein of interest with minimal structural perturbation to the target protein. The aldehyde tag can be inserted at the Nterminus, C-terminus, or internally using standard molecular biology techniques. It can also be inserted at multiple locations at one time to specifically control not only the location but also the number of chemical attachment points. Conjugation of a toxin payload to an aldehyde-containing antibody protein, for example, using aldehyde-targeted conjugation chemistries, results in site-specifically modified proteins (Figure 2B).
ALDEHYDE-TARGETING CONJUGATION CHEMISTRIES
The aldehyde generated by FGE on the target protein can be selectively reacted with alpha-nucleophiles, such as aminooxy- and hydrazide-bearing compounds, generating oxime- and hydrazone-ligated products, respectively. Traditionally, these aldehyde ligation strategies were used because of their chemical simplicity, their bioorthagonality, and because they can be performed under aqueous conditions. However, there are disadvantages to these chemistries. First, the C=N bond formation occurs slowly, and the bioconjugate products are susceptible to hydrolysis, undermining their utility in instances where long-term stability is required. The oxime has been identified as the most hydrolytically stable C=N linkage, but it is still thermodynamically unstable to hydrolysis under dilute conditions. A second disadvantage to oxime formation is the requirement for acidic reaction conditions in the bioconjugation step.12
The ideal bioconjugation reaction would form a stable C-C bond with protein aldehydes under neutral pH conditions. Redwood Bioscience recently described the development of an aldehyde-specific Pictet-Spengler ligation, a C-C bond-forming reaction that capitalizes on the bioorthogonality of hydrazone formation in an intermediate step (Figure 3A). This new ligation strategy, known as hydrazino-Pictet-Spengler (HIPS), utilizes the speed of alpha affect nucleophiles, a hydrazine moiety, to generate an electrophilic intermediate, which is then subject to nucleophilic attack by the indole. The nucleophilic attack forms an irreversible C-C bond, which is responsible for the high stability of this ligation technique. Therefore, HIPS chemistry alleviates the inherent instability associated with the aminooxy- and hydrazine-based aldehyde chemistries. The HIPS reaction forms hydrolytically stable conjugates with glyoxal- and formylglycine-modified proteins, including monoclonal antibodies.13 This proprietary chemistry can be applied to the generation of chemically stable ADCs. In fact, ADCs generated using HIPS chemistry show >90% stability in human plasma after 7 days at 37°C, regardless of the conjugation site (Figure 3B). In addition to producing a very stable C-C bond with aldehydes, the HIPS reaction takes place at neutral or near neutral pH. The ability to generate stable bioconjugates using mild, near physiological pH conditions offers numerous advantages over other existing bioconjugation methods.
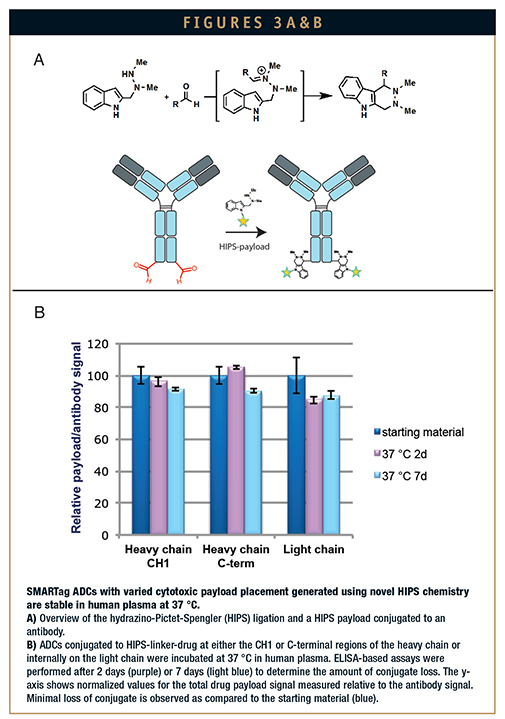
SMARTag technology is a broadly applicable platform that encompasses the use of aldehyde-tagged proteins and HIPS conjugation chemistry. It is a simple and robust system that can be used as a universal scaffold across many fields to generate site-specific, homogenous bioconjugates. Some of the reported applications of the aldehyde tag methodology include fluorophore conjugation, cell surface labeling, PEGylation, glycoengineering, and bispecific protein fusions.7,8,14,15 Redwood Bioscience is specifically interested in applying this proprietary and elegant technology to ADCs.
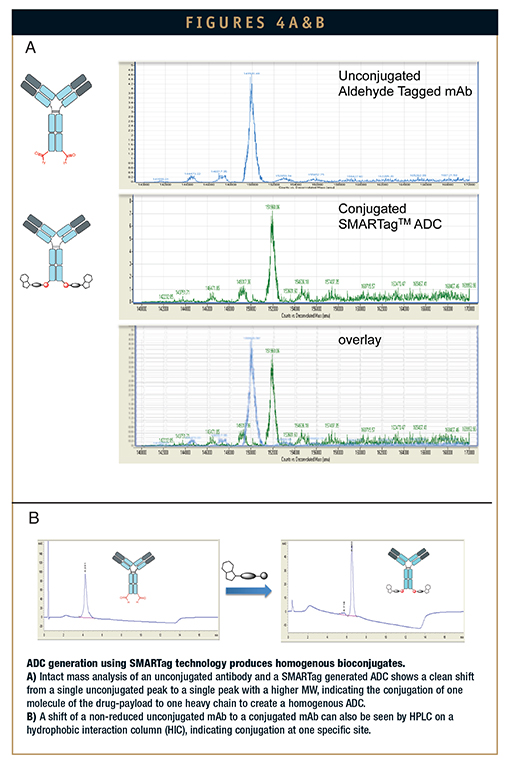
Using HIPS aldehyde chemistry, we can site-specifically modify antibodies at an extensive number of sites by engineering the aldehyde tag into the constant regions of any antibody of interest. HIPS conjugation chemistry applied to payload attachment requires mild, near neutral pH conditions with no refolding required. With respect to payload diversity, the HIPS system is compatible with a variety of payloads that have different cellular targets and modes of action, including microtubule disrupters and DNA alkylators. In addition, Redwood has developed an array of cleavable and non-cleavable linkers utilizing this novel chemistry. Because of the programmable and site-specific nature of HIPS chemistry, the analytics involved in determining the number and location of conjugated drugs to antibodies is significantly simplified relative to traditional, randomly modified bioconjugates (Figure 4). ADCs generated using the SMARTag technology are potent in vitro in the sub-nanomolar range against a variety of targets, using a variety of different payload and linker combinations. Moreover, SMARTag ADCs are efficacious in in vivo mouse tumor models. Together, Redwood Bioscience, Catalent, and SMARTag technology are providing a powerful next-generation solution to the development of optimized ADCs.
To view this issue and all back issues online, please visit www.drug-dev.com.
REFERENCES
1. Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl. 2009;48(38):6974-6998.
2. Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol 2012;30(7):631-637.
3. Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, Senter PD. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem 2008;19(3):759-765.
4. Mueller BM, Wrasidlo WA, Reisfeld RA. Antibody conjugates with morpholinodoxorubicin and acidcleavable linkers. Bioconj Chem. 1990;1(5):325-330.
5. Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925-932.
6. Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;21;20(2):161-167.
7. Rush JS, Bertozzi CR. New aldehyde tag sequences identified by screening formylglycine generating enzymes in vitro and in vivo. J Am Chem Soc. 2008;130(37):12240-12241.
8. Carrico IS, Carlson BL, Bertozzi CR. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3(6):321-322.
9. Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc Natl Acad Sci USA. 2009;106(9):3000-3005.
10. Frese MA, Dierks T. Formylglycine aldehyde Tag-protein engineering through a novel post-translational modification. Chembiochem. 2009;10(3):425-427.
11. Rabuka D, Rush JS, deHart GW, Wu P, Bertozzi CR. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc. 2012;7(6):1052-1067.
12. Rabuka D. Chemoenzymatic methods for site-specific protein modification. Curr Opin Chem Biol. 2010;14(6):790-796.
13. Agarwal P, Kudirka R, Albers AE, Barfield RM, et al. Hydrazino-pictet-spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug Chem. 2013;24(6):846-851.
14. Hudak JE, Yu HH, Bertozzi CR. Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. J Am Chem Soc. 2011;133(40):16127-16135.
15. Hudak JE, Barfield RM, de Hart GW, Grob P, et al. Synthesis of heterobifunctional protein fusions using copper-free click chemistry and the aldehyde tag. Angew Chem Int Ed Engl. 2012;51(17):4161-4165.

Dr. Robyn M. Barfield graduated from the University of Georgia with a BS in Cell Biology and Genetics. She earned her PhD in Cell and Molecular Biology from the University of Pennsylvania, where she studied the Ras signal transduction pathway during Caenorhabditis elegans development in the laboratory of Dr. Meera Sundaram. This work was recognized with an Outstanding Thesis Award. Dr. Barfield’s post-doctoral work focused on the mechanism of protein transport to the cell surface in Saccharomyces cerevisiae under the mentorship of Nobel laureate Dr. Randy Schekman. She currently works as a Senior Research Scientist at Redwood Bioscience as part of the bio-conjugation team that generates site-specific antibody drug conjugates.

Dr. David Rabuka earned his PhD in Chemistry from the University of California, Berkeley, as a Chevron Fellow in the lab of Professor Carolyn Bertozzi. His research included developing and applying Redwood’s platform technology to cell surface modifications. Prior to joining Professor Bertozzi’s lab, Dr. Rabuka worked at the Burnham Institute, synthesizing complex glycans, followed by Optimer Pharmaceuticals, where he was an early employee, focused on the development of glycan and macrolide-based antibiotics. He graduated with a double honors BS in Chemistry and Biochemistry from the University of Saskatchewan, where he earned the Dean’s Science Award and holds an MS in Chemistry from the University of Alberta. Dr. Rabuka is an author on over 20 major publications, as well as numerous book chapters and patents.
Total Page Views: 5649












